
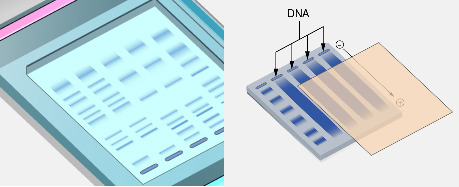
Southern blotting is a classical molecular biology technique used to detect specific sequences of deoxyribonucleic acid (DNA) within a complex mixture of DNA fragments. The method involves transferring DNA fragments separated by gel electrophoresis onto a membrane, followed by hybridization with a labeled probe that specifically binds to the target DNA sequence. This technique allows researchers to identify, analyze, and characterize particular genes or DNA regions within a genome.
The Southern blotting technique was first described in 1975 by the British molecular biologist Sir Edward M. Southern, after whom the method is named. His work introduced a reliable approach for identifying specific nucleotide sequences within large DNA samples, revolutionizing the field of molecular genetics. Since its development, Southern blotting has become an essential laboratory method for gene detection, genome analysis, and the study of genetic variation.
At its core, Southern blotting relies on the principle of nucleic acid hybridization. Complementary DNA strands can bind to each other through base pairing interactions. By using a labeled DNA probe that is complementary to a sequence of interest, scientists can selectively detect that sequence even within a highly complex mixture of DNA fragments. The technique therefore provides a powerful means of locating a specific gene or genetic element among thousands or millions of DNA fragments.
The Southern blotting process typically begins with the extraction and purification of DNA from cells or tissues. The isolated DNA is then digested with restriction enzymes. These enzymes act as molecular scissors, cutting the DNA at specific nucleotide sequences to produce fragments of varying lengths. The resulting DNA fragments are then separated according to size using agarose gel electrophoresis. During electrophoresis, an electric current is applied across the gel, causing negatively charged DNA molecules to migrate toward the positive electrode. Smaller DNA fragments travel faster and therefore move farther through the gel than larger fragments.
Once the DNA fragments have been separated, the next step is to transfer them from the gel onto a solid support membrane, usually made of nitrocellulose or nylon. Before transfer, the DNA within the gel is typically treated with an alkaline solution to denature the double-stranded DNA molecules into single strands. This denaturation step is essential because hybridization probes bind only to single-stranded DNA.
The transfer of DNA fragments from the gel to the membrane is referred to as blotting. In the traditional capillary blotting method, the gel is placed on a buffer-soaked support and covered with the membrane. Layers of absorbent paper towels are placed on top, creating a capillary flow of buffer through the gel. As the buffer moves upward, it carries the DNA fragments out of the gel and onto the membrane, where they bind permanently. After transfer, the DNA fragments retain the same relative positions they occupied in the gel, effectively creating a replica of the electrophoresis pattern on the membrane.
Once the DNA has been immobilized on the membrane, the next step is hybridization with a labeled probe. A probe is a short, single-stranded DNA sequence that is complementary to the DNA region of interest. The probe is labeled so that it can be detected after hybridization. Traditionally, radioactive isotopes such as phosphorus-32 were used to label probes, producing highly sensitive detection signals. However, modern laboratories often use non-radioactive labels such as fluorescent dyes, chemiluminescent molecules, or enzyme-linked markers.
During the hybridization step, the membrane is incubated with the labeled probe under carefully controlled conditions that promote base pairing between complementary DNA strands. If the target DNA sequence is present on the membrane, the probe will bind specifically to that sequence through complementary base pairing. Non-specific binding is minimized through stringent washing steps that remove any probe molecules not firmly bound to the target DNA.
After hybridization and washing, the probe-bound DNA fragments are detected using appropriate imaging techniques. In radioactive detection systems, the membrane is placed against an X-ray film, producing an autoradiograph in which dark bands indicate the presence of the target DNA sequence. In non-radioactive systems, chemiluminescent or fluorescent signals are captured using specialized imaging equipment. The position of the detected band corresponds to the size of the DNA fragment containing the sequence of interest, allowing researchers to analyze genetic structure and variation.
Southern blotting provides several types of information about DNA molecules. It can reveal the presence or absence of specific genes within a genome, determine the size of DNA fragments containing those genes, and identify structural changes such as insertions, deletions, or rearrangements. Because of this capability, the technique has historically played an important role in genetic mapping, gene cloning, and the characterization of genomic organization.
One important application of Southern blotting is in the analysis of gene structure and copy number. By comparing the pattern of DNA fragments detected with a probe, scientists can determine whether a gene exists as a single copy or as multiple copies within a genome. This information is useful in studies of gene duplication, genome evolution, and transgenic organisms. The technique can also be used to confirm the successful insertion of foreign DNA into host genomes during genetic engineering experiments.
Southern blotting has also been widely used in medical genetics. It has been applied to detect mutations associated with genetic disorders, particularly those involving large deletions, insertions, or rearrangements in DNA. For example, before the development of modern sequencing technologies, Southern blot analysis was commonly used to diagnose conditions such as sickle cell anemia and certain forms of thalassemia by identifying specific changes in DNA restriction fragment patterns.
Another significant use of Southern blotting is in forensic science and human identification. The technique played a major role in the early development of DNA fingerprinting methods. By analyzing variations in repetitive DNA sequences known as variable number tandem repeats (VNTRs), scientists could generate unique patterns of DNA fragments for individual persons. These patterns were used for personal identification, paternity testing, and criminal investigations. Although modern forensic laboratories now rely more heavily on polymerase chain reaction (PCR) based methods, Southern blotting laid the foundation for many of these technologies.
Southern blotting has also contributed significantly to evolutionary biology and comparative genomics. By comparing homologous DNA sequences among different species, researchers can investigate evolutionary relationships and genetic conservation. Hybridization patterns observed through Southern blotting provide insights into how genes have been conserved, duplicated, or modified throughout evolutionary history.
Despite its historical importance, Southern blotting has certain limitations. The technique is relatively time-consuming and labor-intensive, often requiring several days to complete. It also requires relatively large amounts of high-quality DNA and careful handling of probes and membranes. Additionally, when radioactive probes are used, special safety precautions and regulatory measures must be followed.
In recent decades, many applications once performed by Southern blotting have been replaced by faster and more sensitive molecular methods such as polymerase chain reaction (PCR), quantitative PCR, and next-generation DNA sequencing. These newer technologies allow rapid detection and characterization of DNA sequences with much smaller sample quantities and higher throughput. Nevertheless, Southern blotting remains a valuable method in certain contexts, particularly when analyzing large genomic rearrangements or verifying transgene integration in genetically modified organisms.
Modern variations of the technique have also been developed to improve efficiency and safety. Non-radioactive labeling systems have largely replaced radioactive probes in many laboratories, reducing health risks and simplifying experimental procedures. Improved membrane materials, hybridization buffers, and detection systems have further enhanced the sensitivity and reliability of the technique.
Southern blotting is a foundational molecular biology technique that enables the detection and analysis of specific DNA sequences within complex mixtures of genetic material. Developed by Sir Edward M. Southern in 1975, the method introduced a powerful approach based on DNA hybridization and membrane transfer. Through a series of steps involving DNA digestion, gel electrophoresis, membrane transfer, probe hybridization, and signal detection, researchers can identify and characterize specific genes or genomic regions. Although newer molecular technologies have expanded the capabilities of genetic analysis, Southern blotting remains an important technique in molecular genetics, forensic science, and genomic research.
Steps in Southern Blotting: Detailed Methodology for DNA Detection and Analysis
Southern blotting is a widely recognized molecular biology technique used for the detection and analysis of specific DNA sequences within complex mixtures of DNA fragments. The method is particularly useful for identifying structural differences between genomes, determining gene organization, and studying related DNA sequences within a genome. Through a combination of DNA digestion, electrophoretic separation, membrane transfer, probe hybridization, and signal detection, Southern blotting enables researchers to locate a particular DNA sequence among thousands of fragments.
Southern blotting remains a fundamental molecular biology technique for the detection and analysis of specific DNA sequences. The method involves a series of carefully coordinated steps, beginning with restriction digestion of DNA and separation of fragments by agarose gel electrophoresis. The DNA fragments are then denatured and transferred onto a nitrocellulose or nylon membrane through capillary blotting. Once immobilized on the membrane, the DNA is hybridized with a labeled probe that specifically binds to the target sequence. Unbound probes are removed through washing, and the hybridized fragments are detected using autoradiography or other imaging methods.
Although newer technologies such as polymerase chain reaction (PCR) and next-generation sequencing have largely replaced Southern blotting for many applications, the technique continues to be valuable in certain areas of genetic research. It provides reliable information about DNA fragment size, gene structure, and genomic organization, making it an important tool in molecular genetics, biomedical research, and forensic science.
Through its ability to detect specific DNA sequences within complex samples, Southern blotting has played a crucial role in advancing our understanding of genetic structure and variation. Even decades after its introduction, it remains an important methodological foundation in molecular biology laboratories around the world.
The technique involves several carefully controlled experimental steps, each of which contributes to the successful detection of the target DNA sequence. These steps include DNA digestion with restriction enzymes, separation of DNA fragments by gel electrophoresis, denaturation of DNA, transfer of DNA fragments to a membrane, hybridization with labeled probes, washing to remove unbound probes, and detection through autoradiography or other imaging techniques. Each step must be performed accurately in order to obtain reliable and interpretable results.
1. Digestion of DNA with Restriction Endonucleases
The first step in Southern blotting is the digestion of genomic DNA using specific restriction endonucleases. Restriction endonucleases are enzymes that recognize and cut DNA at specific nucleotide sequences known as restriction sites. These enzymes function as molecular scissors, cleaving DNA into fragments of varying lengths depending on the distribution of restriction sites within the genome.
The DNA sample to be analyzed is first isolated and purified from the biological material, which may include bacterial cells, plant tissues, animal tissues, or other biological sources. Once purified, the DNA is incubated with one or more restriction enzymes under optimal conditions of temperature, buffer composition, and ionic strength. Each enzyme cuts the DNA at its specific recognition sequence, producing a reproducible set of DNA fragments.
The purpose of restriction digestion is to reduce large DNA molecules into smaller fragments that can be separated and analyzed more easily. The pattern of fragments produced reflects the arrangement of restriction sites in the genome. Differences in DNA sequence between individuals or species may alter these restriction sites, resulting in fragments of different sizes. These variations are often referred to as restriction fragment length polymorphisms (RFLPs), which can be detected through Southern blot analysis.
2. Separation of DNA Fragments by Agarose Gel Electrophoresis
After digestion, the DNA fragments must be separated based on their size. This is accomplished using agarose gel electrophoresis, a common laboratory technique used to resolve DNA fragments of different lengths.
Agarose gel is prepared by dissolving agarose powder in an appropriate buffer solution and heating it until it forms a clear solution. The molten agarose is poured into a gel tray with a comb that creates wells where DNA samples can be loaded. Once the gel solidifies, it forms a porous matrix through which DNA fragments can migrate.
The digested DNA samples are mixed with a loading dye and carefully pipetted into the wells of the gel. An electric field is then applied across the gel. Because DNA molecules carry a negative charge due to their phosphate backbone, they migrate toward the positive electrode when the electric current is applied.
During electrophoresis, smaller DNA fragments move more rapidly through the gel matrix than larger fragments. As a result, the DNA fragments become separated according to their size. After electrophoresis is complete, the gel contains distinct bands representing DNA fragments of different lengths.
At this stage, the DNA fragments are still embedded within the agarose gel. Although the gel allows visualization of the fragments, further processing is required to identify specific DNA sequences.
3. Denaturation of DNA Fragments
Following electrophoresis, the DNA fragments in the gel must be converted from double-stranded DNA (dsDNA) to single-stranded DNA (ssDNA). This step is necessary because hybridization probes can only bind to single-stranded DNA sequences.
The gel slab containing the separated DNA fragments is soaked in an alkaline solution, commonly sodium hydroxide (NaOH). The high pH of this solution disrupts the hydrogen bonds between complementary base pairs, causing the double-stranded DNA molecules to separate into single strands. This process is known as DNA denaturation.
Denaturation serves two important purposes. First, it prepares the DNA fragments for hybridization with complementary probes. Second, it facilitates the transfer of DNA from the gel onto the membrane during the blotting process.
In some protocols, the gel may also undergo a depurination step before denaturation. Depurination involves treating the gel with dilute hydrochloric acid to break very large DNA fragments into smaller pieces, which improves transfer efficiency during blotting.
4. Transfer of DNA Fragments to a Nitrocellulose or Nylon Membrane
Once the DNA fragments have been denatured, they are transferred from the agarose gel onto a solid support membrane. This step is referred to as blotting and is the defining feature of the Southern blot technique.
The membrane used for transfer is usually made of nitrocellulose or nylon. These materials have a strong affinity for nucleic acids and can bind DNA molecules permanently once they come into contact with the membrane surface.
The most traditional method used for transfer is capillary blotting. In this process, the gel is placed on a platform saturated with transfer buffer. The nitrocellulose or nylon membrane is carefully placed on top of the gel, ensuring that no air bubbles are trapped between the gel and the membrane. Several layers of absorbent paper towels are then placed above the membrane.
Buffer moves upward through the gel and membrane by capillary action. As the buffer passes through the gel, it carries the single-stranded DNA fragments along with it. These DNA fragments become trapped and immobilized on the membrane in positions corresponding exactly to their locations within the gel.
This transfer process typically takes several hours to complete, often overnight. By the end of the transfer, the membrane contains a replica of the DNA fragment pattern originally present in the agarose gel.
5. Immobilization of DNA on the Membrane
After transfer, the DNA fragments must be permanently fixed to the membrane so that they remain attached during the subsequent hybridization and washing steps.
For nitrocellulose membranes, immobilization is often achieved by baking the membrane at approximately 80°C in a vacuum oven. For nylon membranes, ultraviolet (UV) crosslinking is commonly used. UV light creates covalent bonds between the DNA molecules and the membrane surface, ensuring that the DNA fragments remain securely attached.
This fixation step stabilizes the DNA and prepares the membrane for probe hybridization.
6. Placement of Weight to Ensure Efficient Transfer
During the blotting process, a stack of absorbent paper towels is placed above the membrane to draw buffer upward through the gel and membrane by capillary action. A weight is often placed on top of the stack to maintain firm contact between the gel, membrane, and absorbent materials.
The weight ensures that the membrane remains evenly pressed against the gel surface, preventing movement or distortion of the DNA bands during transfer. Proper alignment of the layers is critical because any displacement can disrupt the accurate transfer of DNA fragment patterns.
The absorbent paper towels continuously draw buffer through the system, allowing efficient movement of DNA fragments from the gel to the membrane. As the DNA is transferred, it binds strongly to the membrane surface while maintaining the same spatial arrangement that was present in the gel.
7. Hybridization with a Labeled DNA Probe
Once the DNA fragments are immobilized on the membrane, the next step is hybridization with a labeled DNA probe. Hybridization is the process by which a single-stranded probe binds specifically to a complementary DNA sequence present among the immobilized fragments.
The probe used in Southern blotting is typically a short single-stranded DNA molecule designed to match the sequence of the gene or DNA region of interest. Probes may be labeled with radioactive isotopes, fluorescent dyes, or chemiluminescent markers to allow detection after hybridization.
Before hybridization begins, the membrane is often incubated in a prehybridization solution. This solution contains blocking agents that bind to any remaining nonspecific binding sites on the membrane. Prehybridization reduces background signals and improves the specificity of probe binding.
After prehybridization, the labeled probe is added to the hybridization buffer and incubated with the membrane at a controlled temperature. During this incubation period, the probe molecules search for and bind to complementary DNA sequences on the membrane through base pairing interactions between adenine and thymine (A-T) and between guanine and cytosine (G-C).
If the target DNA sequence is present among the immobilized fragments, the probe will hybridize specifically to that sequence, forming a stable double-stranded DNA molecule composed of the probe strand and the target DNA strand.
8. Washing to Remove Unbound Probes
After hybridization is complete, the membrane is washed to remove any probes that have not bound specifically to the target DNA sequence. This washing step is essential for reducing background noise and improving the clarity of the final signal.
The membrane is washed several times using buffer solutions of varying stringency. Stringency refers to the conditions that influence the stability of DNA duplex formation, including temperature, salt concentration, and detergent content.
Under high-stringency conditions, only perfectly complementary DNA strands remain bound together, while mismatched or loosely bound probes are removed. This ensures that the final detected signal corresponds specifically to the DNA sequence of interest.
By the end of the washing step, only probes that are firmly hybridized to complementary DNA fragments remain attached to the membrane.
9. Detection of Hybridized DNA by Autoradiography
The final step in Southern blotting is the detection of the labeled probes that have hybridized to the target DNA fragments. Traditionally, when radioactive probes are used, detection is carried out using autoradiography.
In this method, the membrane is placed in close contact with an X-ray film inside a light-tight cassette. The radioactive isotopes attached to the probe emit radiation that exposes the photographic film. After an appropriate exposure period, the film is developed to reveal dark bands corresponding to the positions of the hybridized DNA fragments (Figure 1).
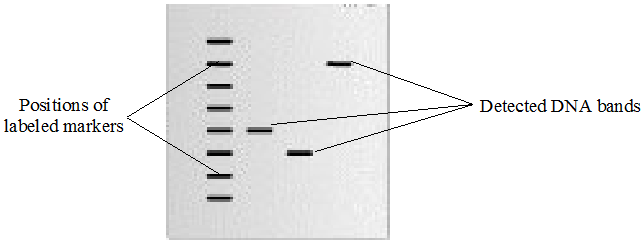
The resulting image is known as an autoradiograph. Each band on the autoradiograph represents a DNA fragment that contains a sequence complementary to the probe. By comparing the positions of these bands with molecular size markers or the original gel pattern, researchers can determine the approximate size of the DNA fragments containing the target sequence.
In modern laboratories, non-radioactive detection systems are often used instead of autoradiography. These systems rely on chemiluminescent or fluorescent signals that can be detected using specialized imaging equipment. Non-radioactive detection methods offer improved safety and faster processing times while still providing high sensitivity.
References
Alberts B, Bray D, Lewis J, Raff M, Roberts K and Watson J.D (2002). The molecular Biology of the Cell. Fourth edition. New York, Garland, USA.
Chen I and Dubnau D (2004). DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2 (3): 241–249.
Cooper G.M and Hausman R.E (2004). The cell: A Molecular Approach. Third edition. ASM Press.
Dale J (2003). Molecular genetics of bacteria. Jeremy W. Dale and Simon Park (4th eds.). John Wiley & Sons Ltd, West Sussex, UK. Pp. 312-313.
Das H.K (2010). Textbook of Biotechnology. Fourth edition. Wiley edition. Wiley India Pvt, Ltd, New Delhi, India.
Lewis R (2004). Human Genetics: Concepts and Applications. Sixth edition. McGraw Hill Publishers, USA.
Lodish H, Berk A, Matsudaira P, Kaiser C.A, Kreiger M, Scott M.P, Zipursky S.L and Darnell J (2004). Molecular Cell Biology. Fifth edition. Scientific American Books, Freeman, New York, USA.
Madigan M.T., Martinko J.M., Dunlap P.V and Clark D.P (2009). Brock Biology of Microorganisms, 12th edition. Pearson Benjamin Cummings Inc, USA.
McPherson M and Moller S (2002). PCR: The Basics. 2nd edition. Taylor and Francis Group. New York, USA.
Sambrook, J., Russell, D.W. (2001). Molecular Cloning: a Laboratory Manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York.
Synder L, Peters J.E, Henkin T.M and Champness W (2013). Molecular Genetics of Bacteria. Fourth edition. American Society of Microbiology Press, USA.
Tamarin Robert H (2002). Principles of Genetics. Seventh edition. Tata McGraw-Hill Publishing Co Ltd, Delhi.
Discover more from Microbiology Class
Subscribe to get the latest posts sent to your email.


