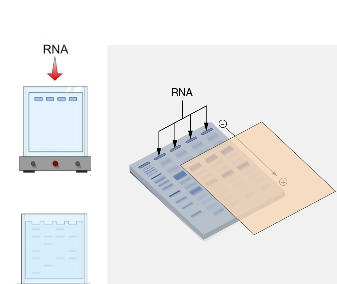
Northern blotting is a classical molecular biology technique used to detect and analyze specific sequences of ribonucleic acid (RNA) within a complex mixture of RNA molecules. It provides valuable information about gene expression by allowing researchers to determine the size, abundance, and integrity of specific RNA transcripts. Although newer technologies such as real-time PCR and RNA sequencing have become widely used for transcript analysis, northern blotting remains an important and reliable method for studying RNA expression patterns and transcript structure.
Northern blotting is a foundational technique in molecular biology that enables the detection and analysis of specific RNA sequences within a complex mixture. Developed in 1977 by James Alwineand colleagues, the method was inspired by the earlier Southern blotting technique introduced by Edwin Southern. While southern blotting detects DNA sequences, northern blotting is specifically designed to identify RNA molecules, particularly messenger RNA, making it a powerful tool for studying gene expression. Through a series of steps involving RNA extraction, electrophoretic separation, membrane transfer, probe hybridization, and signal detection, northern blotting allows researchers to examine both the size and abundance of RNA transcripts. Although modern molecular techniques have introduced faster and more sensitive methods for RNA analysis, northern blotting continues to play an important role in validating gene expression studies and understanding RNA biology.
As aforesaid, the technique was first described in 1977 by James Alwine and his colleagues as a modification of the earlier Edwin Southern DNA detection method. The name “Northern blot” was humorously derived from the earlier Southern blotting technique, even though it has no geographical significance. Instead, the naming convention reflects the lineage of related nucleic acid detection methods. In contrast to southern blotting, which detects specific sequences of DNA, northern blotting is specifically designed to detect RNA molecules, particularly messenger RNA (mRNA), thereby enabling researchers to examine gene expression at the transcriptional level.
Northern blotting operates on the principle of nucleic acid hybridization. In this process, a labeled complementary nucleic acid probe binds specifically to its target RNA sequence through base pairing. When the probe hybridizes with the target RNA immobilized on a membrane, the resulting probe RNA complex can be visualized using autoradiography, fluorescence, or chemiluminescence depending on the labeling system used. This specificity allows researchers to identify and quantify particular RNA transcripts among thousands of different RNA molecules present in a biological sample.
The procedure for northern blotting closely resembles that used in Southern blotting, but it incorporates modifications to accommodate the properties of RNA. The method generally involves several key steps, including RNA extraction, RNA separation by gel electrophoresis, transfer of RNA onto a membrane, hybridization with a labeled probe, and detection of the target RNA sequence.
The first step in northern blotting is the isolation of RNA from biological samples such as tissues, cultured cells, or microorganisms. Because RNA molecules are highly susceptible to degradation by ribonucleases (RNases), strict precautions must be taken during extraction to maintain RNA integrity. Specialized reagents and RNase-free laboratory conditions are commonly used to prevent contamination and degradation. High-quality RNA is essential for accurate results, as degraded RNA may lead to distorted band patterns and unreliable data.
Once RNA is extracted, the molecules are separated based on their size using gel electrophoresis. Typically, denaturing agarose gels containing agents such as formaldehyde are used to ensure that RNA molecules remain linear and do not form secondary structures that could interfere with migration. During electrophoresis, an electric current drives RNA molecules through the gel matrix, causing smaller RNA fragments to migrate faster than larger ones. This step allows researchers to separate RNA transcripts according to their molecular size.
After electrophoresis, the separated RNA molecules must be transferred from the gel onto a solid support membrane. This step is known as blotting and represents the core of the technique. The membrane used is usually made of nylon or nitrocellulose, both of which have a high affinity for nucleic acids. The transfer can be accomplished by capillary action, vacuum transfer, or electroblotting. During this process, RNA molecules move out of the gel and become immobilized on the membrane in the same relative positions they occupied within the gel. Once transferred, the RNA is permanently fixed onto the membrane by baking or ultraviolet (UV) crosslinking.
Following transfer and fixation, the membrane is incubated with a labeled probe that is complementary to the RNA sequence of interest. These probes are typically short single-stranded DNA or RNA molecules that have been labeled with radioactive isotopes, fluorescent dyes, or chemiluminescent compounds. The probe hybridizes specifically to the target RNA through complementary base pairing. Before hybridization, the membrane is often treated with a blocking solution to reduce nonspecific binding and improve the specificity of probe interaction.
Hybridization is carried out under controlled conditions of temperature, salt concentration, and pH to promote the formation of stable probe–target complexes. After sufficient incubation time, the membrane undergoes a series of washing steps designed to remove unbound or nonspecifically bound probes. These washes are performed under increasingly stringent conditions to ensure that only probes perfectly complementary to the target RNA remain bound.
The final step in northern blotting is the detection of the hybridized probe. The method used depends on the labeling system incorporated into the probe. In traditional northern blotting, radioactive probes were commonly used, and detection involved exposing the membrane to X-ray film to produce an autoradiograph. In modern laboratories, non-radioactive labeling systems such as fluorescent or chemiluminescent probes are frequently used due to safety and convenience. The resulting signal appears as distinct bands on the detection medium, corresponding to the size and quantity of the RNA transcript of interest.
One of the major advantages of northern blotting is its ability to provide information about the size of RNA transcripts. Unlike some modern transcript quantification methods, northern blotting allows researchers to verify whether the detected RNA corresponds to the expected transcript length. This is particularly useful for identifying alternative splicing events, RNA processing intermediates, or degraded RNA products. The intensity of the detected band also reflects the abundance of the RNA molecule, allowing for semi-quantitative comparisons of gene expression levels between samples.
Northern blotting has been widely used in gene expression studies across many biological disciplines. Researchers employ this technique to investigate how genes are regulated during development, disease, or environmental stress. By analyzing RNA expression patterns, scientists can determine when and where specific genes are active within cells or tissues. The method is also useful for validating results obtained from other high-throughput techniques such as microarray analysis or RNA sequencing.
Despite its usefulness, northern blotting has several limitations. The technique requires relatively large amounts of high-quality RNA compared to more sensitive methods like reverse transcription polymerase chain reaction (RT-PCR). In addition, the procedure can be time-consuming and labor-intensive, often requiring several days to complete from RNA extraction to final detection. The use of radioactive probes in traditional protocols also raises safety and disposal concerns, although these issues have been mitigated by the adoption of non-radioactive labeling systems.
Another limitation is that northern blotting provides lower sensitivity than some modern molecular techniques. For example, very low-abundance transcripts may be difficult to detect without amplification steps. However, its reliability and ability to confirm transcript size continue to make it a valuable technique in molecular biology laboratories. For these reasons, northern blotting is often used as a complementary method alongside newer technologies rather than being completely replaced by them.
Over the years, several variations and improvements to the original northern blotting protocol have been developed. These modifications include the use of different membrane materials, enhanced probe labeling strategies, and automated detection systems. Such advancements have improved the sensitivity, speed, and safety of the technique while maintaining its fundamental principle of nucleic acid hybridization.
Steps in Northern Blotting: Detailed Procedure for RNA Detection and Analysis
Northern blotting is a widely used molecular biology technique for the detection and analysis of specific RNA molecules within a complex mixture. The method enables researchers to determine both the size and the abundance of RNA transcripts, particularly messenger RNA (mRNA), which reflects gene expression in cells and tissues. The technique was first described in 1977 by James Alwine and colleagues as an adaptation of the DNA detection method developed by Edwin Southern. While Southern blotting is designed to detect DNA sequences, northern blotting specifically detects RNA molecules using complementary nucleic acid probes.
The northern blotting procedure involves several sequential steps: RNA isolation, electrophoretic separation of RNA molecules, transfer of RNA onto a membrane, hybridization with a labeled probe, washing to remove nonspecific binding, and detection of the hybridized RNA-probe complexes. Each step must be carried out carefully to ensure the integrity of the RNA and the accuracy of the results.
1. Isolation and Preparation of RNA
The first step in northern blotting is the isolation of RNA from biological samples such as tissues, cultured cells, or microorganisms. RNA extraction must be performed under conditions that prevent degradation by ribonucleases (RNases), which are enzymes that rapidly break down RNA molecules. Laboratory reagents, glassware, and plasticware used during RNA isolation must therefore be RNase-free. Chemical reagents and specialized protocols are often employed to protect RNA molecules and ensure that high-quality RNA is obtained.
Once the RNA has been isolated, its concentration and integrity are typically assessed using spectrophotometry and electrophoresis. Intact RNA is essential for reliable northern blotting because degraded RNA can lead to inaccurate band patterns and misinterpretation of gene expression levels.
In some protocols, RNA samples may undergo enzymatic treatment to remove contaminating DNA. Although restriction endonucleases are widely used in DNA analysis, RNA samples in northern blotting are generally not digested with restriction enzymes. Instead, the RNA is maintained in its natural form so that the full length of transcripts can be analyzed. Preserving intact RNA allows researchers to determine transcript size and identify different RNA isoforms.
2. Denaturation and Preparation of RNA Samples
Before electrophoresis, RNA samples are usually treated with denaturing agents such as formaldehyde or glyoxal. These chemicals disrupt hydrogen bonding within RNA molecules, preventing the formation of secondary structures such as hairpins or loops. Without denaturation, RNA molecules might fold into complex shapes that would affect their migration through the gel.
The denatured RNA is then mixed with a loading buffer that contains dyes and density agents. The dyes help track the progress of electrophoresis, while the density agents ensure that the RNA samples sink properly into the wells of the gel.
3. Separation of RNA by Agarose Gel Electrophoresis
After preparation, the RNA samples are loaded onto an agarose gel for electrophoresis. Agarose gel electrophoresis is a standard laboratory technique used to separate nucleic acids according to their molecular size. The gel contains a denaturing agent, usually formaldehyde, which keeps the RNA molecules in a linear configuration during the separation process.
An electric current is applied across the gel, causing negatively charged RNA molecules to migrate toward the positive electrode. Smaller RNA fragments move more rapidly through the gel matrix, whereas larger molecules migrate more slowly. As a result, RNA molecules become separated according to their size.
This separation step is crucial because it allows researchers to distinguish between RNA transcripts of different lengths. After electrophoresis, the RNA molecules remain distributed within the gel in distinct bands corresponding to their molecular sizes.
4. Treatment of the Gel and Preparation for Transfer
Following electrophoresis, the gel containing the separated RNA molecules must be prepared for transfer to a membrane. In some protocols, the gel is briefly treated with solutions that facilitate the transfer process and improve RNA binding to the membrane. For example, soaking the gel in an alkaline solution such as sodium hydroxide (NaOH) can help denature RNA molecules and make them more accessible for hybridization with complementary probes.
Neutralizing buffers may then be applied to stabilize the RNA molecules and prepare them for transfer. This step ensures that the RNA remains single-stranded and capable of forming complementary base pairs with the probe during hybridization.
5. Transfer of RNA from Gel to Membrane (Blotting)
The next step is the transfer of RNA molecules from the agarose gel onto a solid membrane support. This process is known as blotting and is the defining feature of the technique.
Traditionally, nitrocellulose membranes were used for this purpose, although nylon membranes are now commonly employed due to their greater durability and higher binding capacity for nucleic acids. The membrane acts as an absorbent surface onto which RNA molecules become immobilized.
The transfer is typically achieved through capillary action. In this process, the agarose gel is placed on top of a stack of absorbent paper soaked in transfer buffer. The nitrocellulose or nylon membrane is carefully placed on top of the gel, followed by additional layers of absorbent paper and a weight to maintain contact between the layers.
As the transfer buffer moves upward through the stack by capillary action, it carries the RNA molecules out of the gel and onto the membrane. During this process, the RNA fragments become bound to the membrane in positions corresponding exactly to their locations within the gel. In other words, the spatial arrangement of RNA bands is preserved during transfer.
Once the transfer is complete, the membrane is usually baked at a high temperature or exposed to ultraviolet light to permanently fix the RNA molecules onto its surface. This fixation step ensures that the RNA remains attached to the membrane during subsequent washing and hybridization steps.
6. Pre-Hybridization of the Membrane
Before adding the labeled probe, the membrane is often incubated in a pre-hybridization buffer. This buffer contains blocking agents such as denatured DNA, proteins, or detergents that coat the membrane and reduce nonspecific binding of the probe.
Pre-hybridization improves the specificity of the hybridization reaction by ensuring that the labeled probe binds only to the complementary RNA sequence rather than randomly attaching to other parts of the membrane.
7. Hybridization with a Labeled Probe
After pre-hybridization, the membrane is placed in a dish containing hybridization buffer along with a labeled nucleic acid probe designed to recognize the RNA sequence of interest. The probe is usually a short strand of DNA or RNA whose sequence is complementary to the target RNA transcript.
Traditionally, probes were labeled with radioactive isotopes such as phosphorus-32. These radioactive probes produce highly sensitive signals that can be detected using X-ray film. In modern laboratories, however, non-radioactive labeling methods using fluorescent dyes or chemiluminescent compounds are increasingly preferred due to safety and environmental considerations.
During hybridization, the probe binds specifically to its complementary RNA sequence through base pairing. This process typically occurs at controlled temperatures that promote stable hybrid formation while minimizing nonspecific interactions.
Hybridization may last several hours or overnight to allow sufficient time for the probe to locate and bind to its target RNA molecules on the membrane.
8. Washing of the Membrane
After hybridization, the membrane undergoes a series of washing steps designed to remove any probe molecules that are not specifically bound to the target RNA. These washes are performed using buffers of increasing stringency, meaning that the conditions become progressively more selective for perfectly matched probe-target pairs.
The washing process is essential for reducing background noise and ensuring that only the correctly hybridized probe molecules remain attached to the membrane. This improves the clarity and reliability of the final detection results.
9. Detection of Hybridized RNA–Probe Complexes
Once washing is complete, the membrane is ready for detection of the hybridized RNA-probe complexes. If radioactive probes are used, the membrane is placed against an X-ray film in a process known as autoradiography. The radioactive emissions from the probe expose the film, producing dark bands that correspond to the locations of the target RNA molecules.
After an appropriate exposure period, the film is developed to reveal the band pattern. Each band represents a specific RNA transcript that has hybridized with the probe.
When non-radioactive probes are used, detection may involve fluorescence imaging or chemiluminescent reactions that produce light signals captured by imaging equipment.
10. Analysis and Interpretation of Results
The final step in northern blotting involves analyzing the band patterns obtained from the detection process. The position of each band indicates the size of the RNA transcript, while the intensity of the band reflects the relative abundance of that transcript in the sample.
Researchers often compare the expression patterns of the RNA sequence of interest across multiple samples, such as different tissues, developmental stages, or experimental treatments. By examining variations in band intensity, scientists can determine whether a gene is upregulated, downregulated, or expressed at constant levels under different conditions.
For accurate interpretation, northern blot results are often normalized using reference genes or housekeeping genes whose expression levels remain relatively stable across samples. This normalization allows for meaningful comparisons between experimental groups.
Northern blotting remains a fundamental technique for studying RNA expression and transcript structure. The method involves isolating RNA, separating it by agarose gel electrophoresis, transferring the RNA onto a membrane, hybridizing it with a labeled probe, and detecting the resulting probe-RNA complexes. Through these steps, researchers can determine both the size and abundance of specific RNA transcripts within biological samples.
Although modern molecular techniques such as quantitative PCR and RNA sequencing provide greater sensitivity and throughput, northern blotting continues to be widely used because of its reliability and its ability to reveal transcript size and integrity. For this reason, it remains an important tool in molecular biology for validating gene expression data and investigating the regulation of gene activity at the RNA level.
References
Alberts B, Bray D, Lewis J, Raff M, Roberts K and Watson J.D (2002). The molecular Biology of the Cell. Fourth edition. New York, Garland, USA.
Chen I and Dubnau D (2004). DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2 (3): 241–249.
Cooper G.M and Hausman R.E (2004). The cell: A Molecular Approach. Third edition. ASM Press.
Dale J (2003). Molecular genetics of bacteria. Jeremy W. Dale and Simon Park (4th eds.). John Wiley & Sons Ltd, West Sussex, UK. Pp. 312-313.
Das H.K (2010). Textbook of Biotechnology. Fourth edition. Wiley edition. Wiley India Pvt, Ltd, New Delhi, India.
Lewis R (2004). Human Genetics: Concepts and Applications. Sixth edition. McGraw Hill Publishers, USA.
Lodish H, Berk A, Matsudaira P, Kaiser C.A, Kreiger M, Scott M.P, Zipursky S.L and Darnell J (2004). Molecular Cell Biology. Fifth edition. Scientific American Books, Freeman, New York, USA.
Madigan M.T., Martinko J.M., Dunlap P.V and Clark D.P (2009). Brock Biology of Microorganisms, 12th edition. Pearson Benjamin Cummings Inc, USA.
McPherson M and Moller S (2002). PCR: The Basics. 2nd edition. Taylor and Francis Group. New York, USA.
Sambrook, J., Russell, D.W. (2001). Molecular Cloning: a Laboratory Manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York.
Synder L, Peters J.E, Henkin T.M and Champness W (2013). Molecular Genetics of Bacteria. Fourth edition. American Society of Microbiology Press, USA.
Tamarin Robert H (2002). Principles of Genetics. Seventh edition. Tata McGraw-Hill Publishing Co Ltd, Delhi.
Discover more from Microbiology Class
Subscribe to get the latest posts sent to your email.