Prions are unique sub-viral infectious agents composed primarily of proteins. Unlike conventional infectious organisms such as bacteria, fungi, parasites, and viruses, prions lack nucleic acids, including deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). Their unusual composition and mode of infection distinguish them from all other known pathogens. Prions are therefore regarded as infectious proteins capable of transmitting disease in both humans and animals without the involvement of genetic material.
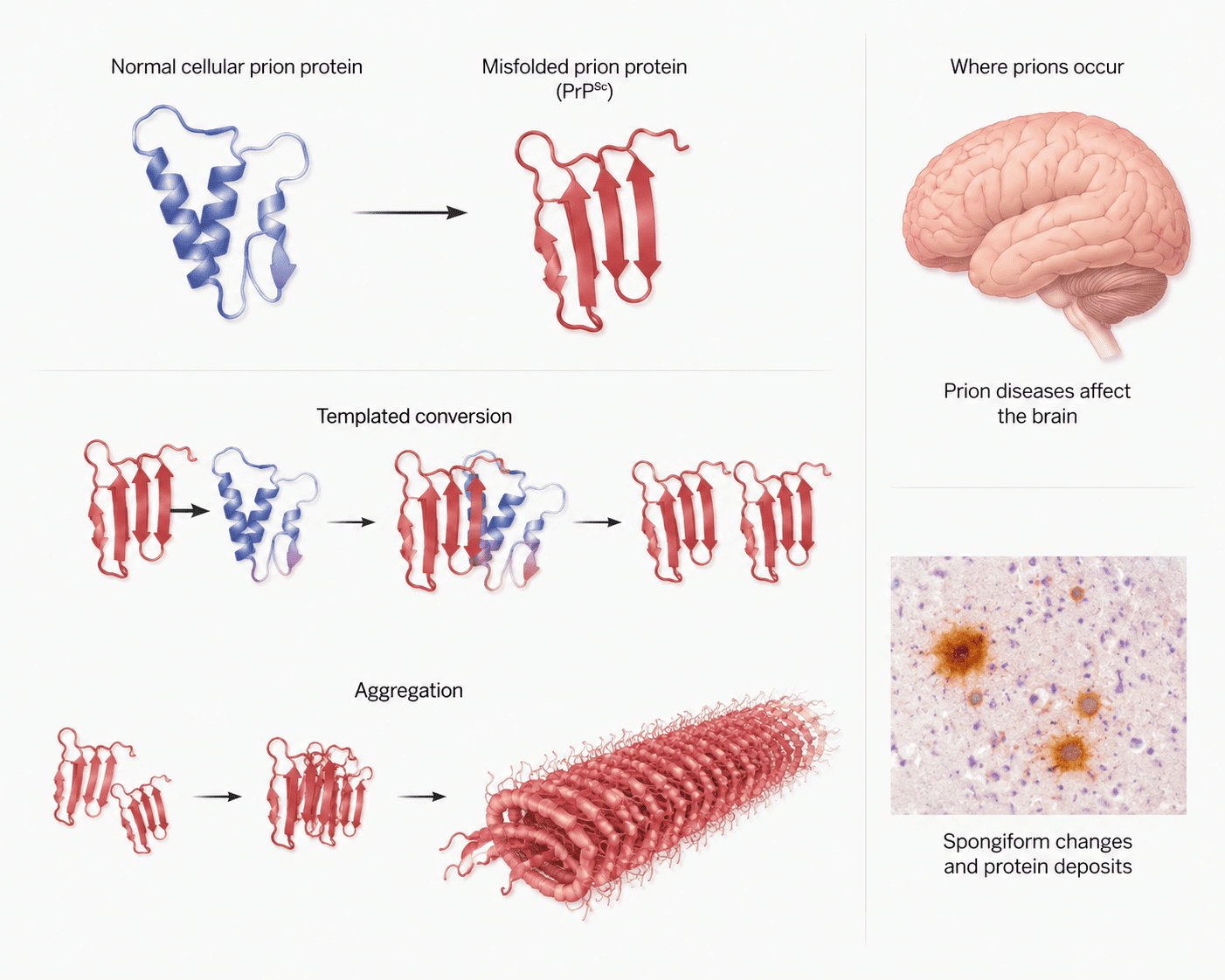
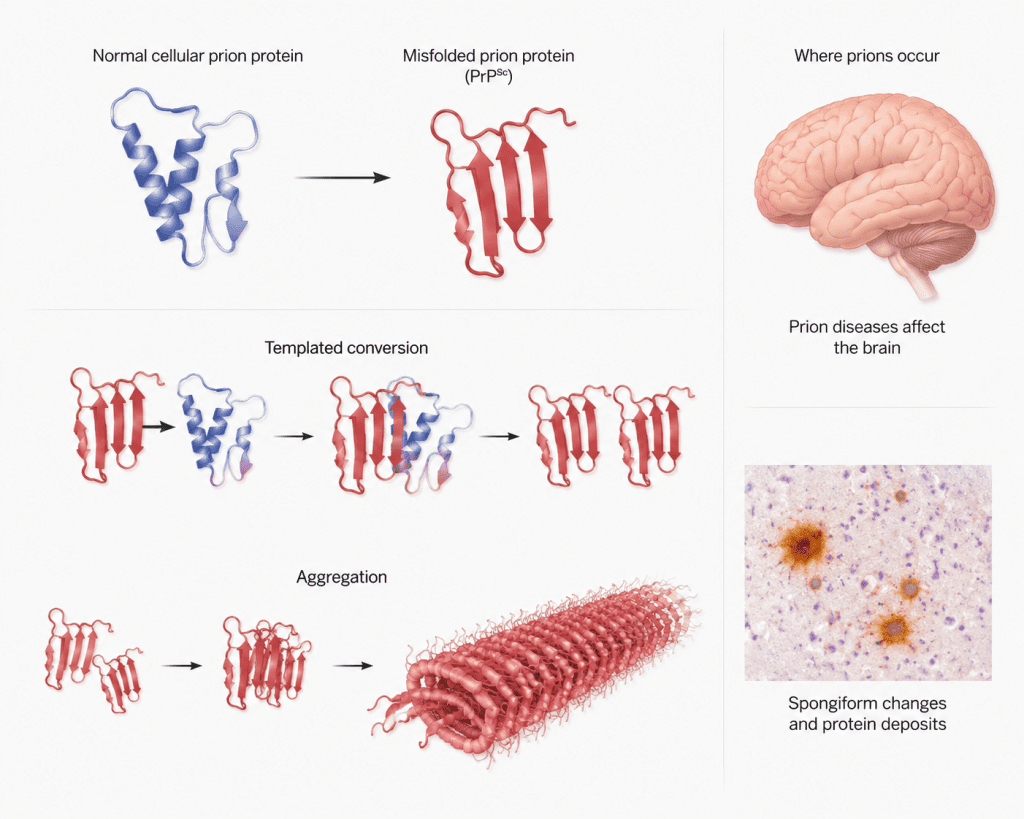
The term “prion” is derived from the phrase “proteinaceous infectious particle.” These infectious agents are mainly composed of an abnormal and pathogenic isoform of a naturally occurring cellular protein known as the prion protein (PrP). The normal form, referred to as PrPᶜ, is harmless and is commonly found in the nervous tissues of mammals. However, when this normal protein undergoes abnormal folding, it transforms into a disease-causing form called PrPˢᶜ. This abnormal isoform possesses the ability to convert normal proteins into similarly misfolded forms, thereby propagating infection within the host.
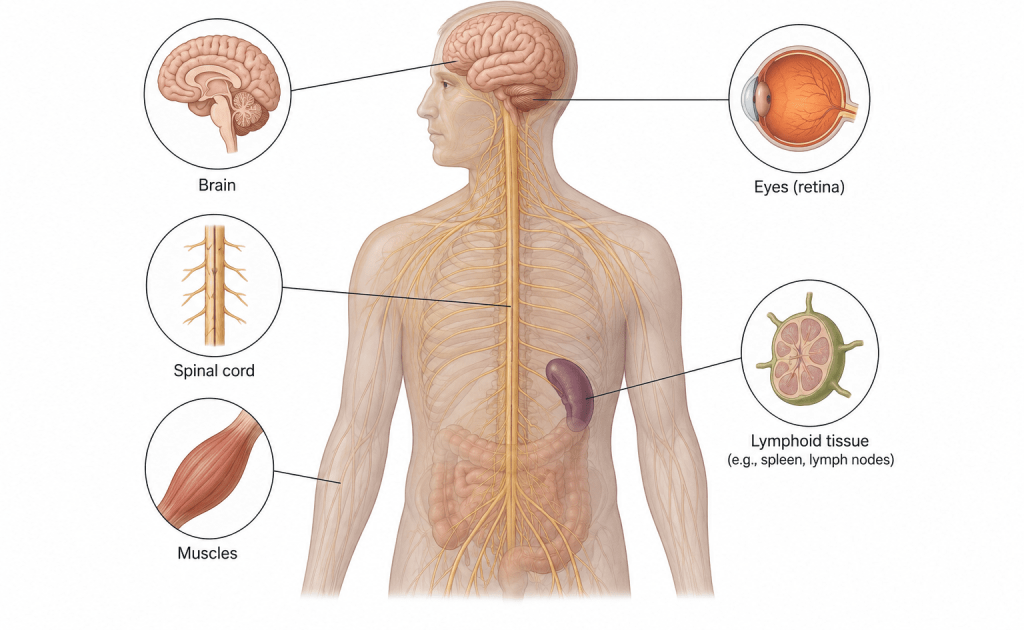
Prions are responsible for a group of fatal neurodegenerative disorders collectively known as transmissible spongiform encephalopathies (TSEs). In humans, these diseases include Creutzfeldt-Jakob disease (CJD), kuru, Gerstmann–Sträussler–Scheinker syndrome, and fatal familial insomnia. In animals, prion diseases include bovine spongiform encephalopathy (BSE), commonly known as mad cow disease, scrapie in sheep and goats, and chronic wasting disease in deer and elk. These diseases are characterized by progressive degeneration of brain tissue, resulting in memory loss, impaired coordination, dementia, and eventually death.
Prions differ significantly from viruses in several important ways. Viruses contain either DNA or RNA enclosed within a protein coat called a capsid, and some viruses also possess an outer lipid envelope. Their replication depends on the genetic information encoded within their nucleic acids. In contrast, prions are completely devoid of nucleic acids and replicate through protein misfolding mechanisms rather than genetic replication. This makes prions fundamentally different from conventional virions.
Another major distinction between prions and viruses lies in their interaction with the immune system. Viruses are generally immunogenic, meaning they stimulate the host immune system to produce antibodies and other defensive responses. Prions, however, are largely non-immunogenic because the pathogenic prion protein closely resembles the normal proteins naturally present in the host. As a result, the mammalian immune system is often unable to recognize pathogenic prions as foreign agents and therefore fails to mount an effective immune response against them.
Prions exhibit remarkable resistance to physical and chemical agents that typically destroy viruses and bacteria. They are resistant to heat, radiation, ultraviolet light, and many disinfectants, making them particularly difficult to eliminate in medical and laboratory settings. Their resistance contributes significantly to their persistence and transmissibility.
Prions are highly unusual infectious agents composed solely of abnormal proteins without any DNA or RNA. Their unique mechanism of infection, resistance to immune detection, and ability to cause fatal neurodegenerative diseases clearly distinguish them from viruses and other pathogens.
Pathogenic and cellular forms of prion proteins
The major identifiable macromolecules associated with prions are known as prion protein scrapie (PrPSc). These proteins represent the abnormal and pathogenic forms of prion proteins that are responsible for the development and transmission of prion diseases in humans and animals. The name “scrapie” was derived from scrapie disease, a fatal neurodegenerative disorder observed in sheep and goats. PrPSc possesses an altered three-dimensional structure that distinguishes it from the normal cellular form of the protein. This abnormal configuration makes the pathogenic protein highly resistant to enzymatic digestion, heat, and chemical agents, allowing it to accumulate within nervous tissues, especially in the brain.
The normal and naturally occurring form of the prion protein is called prion protein cellular (PrPC). This cellular isoform is commonly found in mammalian cells, particularly in neurons and other tissues of the central nervous system. Unlike the pathogenic form, PrPC is harmless, soluble, and easily degraded by cellular enzymes. Scientists believe that PrPC plays important physiological roles in cellular communication, signal transduction, neuronal survival, and maintenance of normal brain function.
Mammalian prions reproduce through a highly unusual mechanism that does not involve nucleic acids such as DNA or RNA. Instead of replicating genetically like viruses or bacteria, pathogenic PrPSc propagates by interacting directly with the normal PrPC proteins. During this interaction, PrPSc induces structural changes in the normal proteins, converting them into additional pathogenic forms. This process of protein misfolding creates a chain reaction in which increasing amounts of abnormal proteins accumulate within brain tissues.
The accumulation of PrPSc in neural tissues eventually leads to severe neurodegeneration, brain damage, and the formation of sponge-like holes in brain tissue. These pathological changes are characteristic features of transmissible spongiform encephalopathies, including Creutzfeldt-Jakob disease in humans and bovine spongiform encephalopathy in cattle.
Though they lack nucleic acids (which are unique in carrying genetic information for disease initiation in a particular host), the prions cause diseases in animals and mammals as aforementioned. Kuru disease (that occur in cannibalistic mammals), Creutzfeldt- Jakob disease (CJD) that occur in humans, scrapie (that occurs in goat and sheep), bovine spongiform encephalopathy (BSE) that occur in cattle and chronic wasting disease (that occur in deer and elk) amongst others are typical examples of prion diseases in animals and mammals (Table 1).
Transmissible spongiform encephalopathy (TSE) is the fatal prion disease of the nervous system that is characterized by the degeneration of the spongiform of the brain in animals. Animal prion diseases are collectively known as transmissible spongiform encephalopathies (TSEs). Since prions lack nucleic acid molecules, how then do they replicate in vivo to initiate a disease process or produce their own progeny from parental cells? Prion molecules as aforementioned are mainly made up of PrP; and the host cell usually contains a gene that encodes the native form of the prion proteins (i.e. PrPC).This gene in the host cell that encodes for PrPC is known as the human PrP gene (Prnp); and the gene is located in chromosome number 20 in humans and chromosome number 2 in mice.
Table 1. Summary of some prion diseases in man and animals
| Prion disease | Natural host |
| Kuru | Humans |
| Scrapie | Sheep and goat |
| Creutzfeldt-Jakob disease (CJD) | Humans |
| Chronic wasting disease | Elk and deer |
| Fatal familial insomnia (FFI) | Humans |
| Bovine spongiform encephalopathy (BSE) | Cattles |
| Fatal sporadic insomnia (FSI) | Humans |
| Feline spongiform encephalopathy (FSE) | Cats |
| Gerstmann-Straussler-Scheinker syndrome (GSSS) | Humans |
Pathogenesis of prion
Prion pathogenesis refers to the process through which pathogenic prion proteins invade, multiply, and cause disease in humans and animals (Figure 1). The process begins when the abnormal prion protein, known as PrPSc, enters the body through infected food, contaminated medical instruments, inherited genetic mutations, or spontaneous protein misfolding. Unlike conventional pathogens, prions do not contain DNA or RNA; instead, their infectivity depends entirely on their abnormal protein structure. After entering the host, PrPSc interacts with the normal cellular prion protein, PrPC, which is commonly present in nerve cells and other tissues. The pathogenic prion protein acts as a template, inducing the normal PrPC proteins to change their structure into abnormal PrPSc forms. This conversion process leads to the gradual accumulation of insoluble prion aggregates within tissues, particularly in the central nervous system.
As PrPSc accumulates in the brain, it causes progressive neuronal degeneration, vacuolation, and the formation of sponge-like holes in brain tissue. These pathological changes interfere with normal brain function and result in severe neurological symptoms such as memory loss, behavioral abnormalities, impaired coordination, tremors, and dementia. Prion diseases are usually fatal because the damage to nervous tissue is irreversible. The incubation period of prion diseases may last for several years before clinical symptoms appear. Once symptoms develop, the disease progresses rapidly and eventually leads to death.
The pathogenic form of prion proteins (i.e. PrPSc) is identical to the cellular isoform of the prion protein (PrPC), which is non-pathogenic in nature. This genetic similarity between the epitope of PrPC and PrPSc is responsible for the non-immunogenicity of prions, and the reason why host cells fails to mount an immune response to prion invasion the same it mounts an attack against viral agents that enter the body.
The invasion of a host cell by PrPSc (pathogenic prion protein) stimulates the genetic conversion of PrPC (the normal and non-infectious prion protein)to PrPSc; and this is particularly applicable in host cells that are expressing PrPC. PrPC and PrPSc are the two known conformations in which prion proteins exist. It is however noteworthy that the replication of prions in host cells is generally facilitated when the sequences of the PrPC and PrPSc are identical.
Thus the presence of PrPC is vital for the development of prion diseases in mammals and animals; and this has been confirmed in knock-out mice (i.e. mice whose Prnp gene have been genetically modified) which are unable to form the PrPC proteins and the prion disease after several challenges with PrPSc, the pathogenic prion proteins. A pathogenic prion protein (PrPSc) replicates itself in host cells be converting healthy prion proteins (PrPC) to the pathogenic isoforms (i.e. PrPSc). PrPCisnormally produced by cells of the neurons in mammals and animals; and a misfolding of the PrPC during its synthesis in the neuron can lead to the conversion of PrPC to PrPSc which is insoluble and resistant to proteases (protein degrading enzymes).
The formation of PrPSc leads to the development of aggregates of the pathogenic prion proteins, and this phenomenon causes a damage of the neural tissues, brain tissues and other neurological symptoms that causes prion diseases to ensue. Profound neurological dysfunction in animal and mammalian hosts infected by pathogenic prion proteins is usually fatal in nature. Prion diseases are generally initiated in host cells by a molecular mechanism that causes pathogenic prion proteins to arise spontaneously; and infectious prion proteins are infectious and can be transferred from one infectious host to another susceptible host.
Prions have also been discovered to infect fungi especially yeast cells aside animals and mammalian cells that they are mainly known to infect. There is currently no therapeutic option for the treatment of prion diseases even though several compounds have been tested and suggested for use in the management of neurodegenerative diseases caused by pathogenic prion proteins.
Prion protein misfolding and neurodegeneration pathway
Prion protein biology is framed through a progression from native cellular conformations to pathogenic misfolded assemblies, emphasizing structural instability as a driver of disease (Figure 2). Normal prion protein (PrPC) adopts an alpha-helical rich structure, whereas the pathogenic isoform (PrPSc) shifts toward beta-sheet dominance, enabling self-propagating templating. Conversion dynamics highlight how misfolded species recruit native proteins, amplifying conformational change through autocatalytic cycles. Subsequent aggregation leads to fibrillar assemblies that accumulate in neural tissue, correlating with spongiform degeneration.
Spatial context links these molecular events to brain involvement, where protein deposits associate with neuronal loss and vacuolation. Histological patterns reinforce the relationship between aggregate burden and tissue disruption. The progression from molecular misfolding to macroscopic neurodegeneration underscores the continuity between structural biology and neuropathology. Prion-driven mechanisms integrate protein chemistry, kinetic propagation, and tissue-level pathology into a unified disease model of transmissible protein misfolding, offering conceptual clarity for disease mechanism understanding across biological scales.
Detection and treatment of prion diseases
The detection of prion diseases is often difficult because the symptoms usually resemble those of other neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease. Diagnosis commonly involves a combination of clinical examination, laboratory analysis, and imaging techniques. Neurological symptoms such as dementia, memory loss, muscle stiffness, impaired coordination, and behavioral changes may raise suspicion of prion infection. Magnetic resonance imaging (MRI) and electroencephalography (EEG) are frequently used to identify abnormal brain activity and structural damage associated with prion diseases.
Laboratory tests may include examination of cerebrospinal fluid for specific protein markers linked to neuronal degeneration. Advanced molecular techniques such as real-time quaking-induced conversion (RT-QuIC) have improved the early detection of pathogenic prion proteins with high sensitivity and specificity. Definitive diagnosis is usually confirmed through brain biopsy or postmortem examination, where sponge-like degeneration and accumulation of abnormal prion proteins can be observed in nervous tissues.
There is no effective cure or specific treatment for prion diseases. Available treatments are mainly supportive and aimed at relieving symptoms, reducing discomfort, and improving the quality of life of affected individuals. Medications may be administered to control pain, muscle spasms, anxiety, and sleep disturbances. Prevention remains the most effective control strategy and includes strict sterilization procedures, screening of animal products, and avoidance of contaminated tissues or medical instruments.
Control and prevention of prion diseases
Control and prevention of prion diseases rely primarily on interrupting transmission pathways, as no curative treatment currently exists. Because prions are highly resistant to conventional disinfection methods, strict biosafety and biosecurity measures are essential in healthcare, laboratory, and agricultural settings. In medical environments, prevention focuses on avoiding iatrogenic transmission, which can occur through contaminated surgical instruments, tissue grafts, or biological products. Standard sterilization procedures are often insufficient against prions; therefore, enhanced decontamination protocols are required. These may include extended autoclaving at higher temperatures and the use of strong chemical agents such as sodium hypochlorite or sodium hydroxide. Dedicated surgical instruments for high-risk procedures are also recommended to minimize cross-contamination.
In public health and food safety, control measures include surveillance of livestock diseases such as bovine spongiform encephalopathy (BSE) and strict regulation of animal feed to prevent recycling of infected tissues. Removal of specified risk materials, such as brain and spinal cord tissues from slaughtered animals, reduces the risk of human exposure through the food chain. Monitoring programs and rapid reporting systems are essential for early detection of outbreaks. Genetic counseling may be useful in families with inherited prion diseases, as certain mutations in the prion protein gene increase susceptibility. In addition, public education plays a key role in reducing exposure risks and promoting safe handling of animal products. Effective control of prion diseases depends on rigorous infection control practices, continuous surveillance, and strict adherence to food safety regulations.
References
Acheson N.H (2011). Fundamentals of Molecular Virology. Second edition. John Wiley and Sons Limited, West Sussex, United Kingdom.
Alan J. Cann (2005). Principles of Molecular Virology. 4th edition. Elsevier Academic Press, Burlington, MA, USA.
Alberts B, Bray D, Johnson A, Lewis J, Raff M, Roberts K and Walter P (1998). Essential Cell Biology: An Introduction to the Molecular Biology of the Cell. Third edition. Garland Publishing Inc., New York.
Barrett J.T (1998). Microbiology and Immunology Concepts. Philadelphia, PA: Lippincott-Raven Publishers. USA.
Black, J.G. (2008). Microbiology: Principles and Explorations (7th ed.). Hoboken, NJ: J. Wiley & Sons.
Brian W.J Mahy and Mark H.C van Regenmortel (2010). Desk Encyclopedia of Human and Medical Virology. Elsevier Academic Press, San Diego, USA.
Brooks G.F., Butel J.S and Morse S.A (2004). Medical Microbiology, 23rd edition. McGraw Hill Publishers. USA.
Cann A.J (2011). Principles of Molecular Virology. Fifth edition. Academic Press, San Diego, United States.
Carter J and Saunders V (2013). Virology: Principles and Applications. Second edition. Wiley-Blackwell, New Jersey, United States.
Champoux J.J, Neidhardt F.C, Drew W.L and Plorde J.J (2004). Sherris Medical Microbiology: An Introduction to Infectious Diseases. 4th edition. McGraw Hill Companies Inc, USA.
Dimmock N (2015). Introduction to Modern Virology. Seventh edition. Wiley-Blackwell, New Jersey, United States.
Dimmock N.J, Easton A.J and Leppard K.N (2001). Introduction to modern virology. 5th edition. Blackwell Science publishers. Oxford, UK.
Discover more from Microbiology Class
Subscribe to get the latest posts sent to your email.