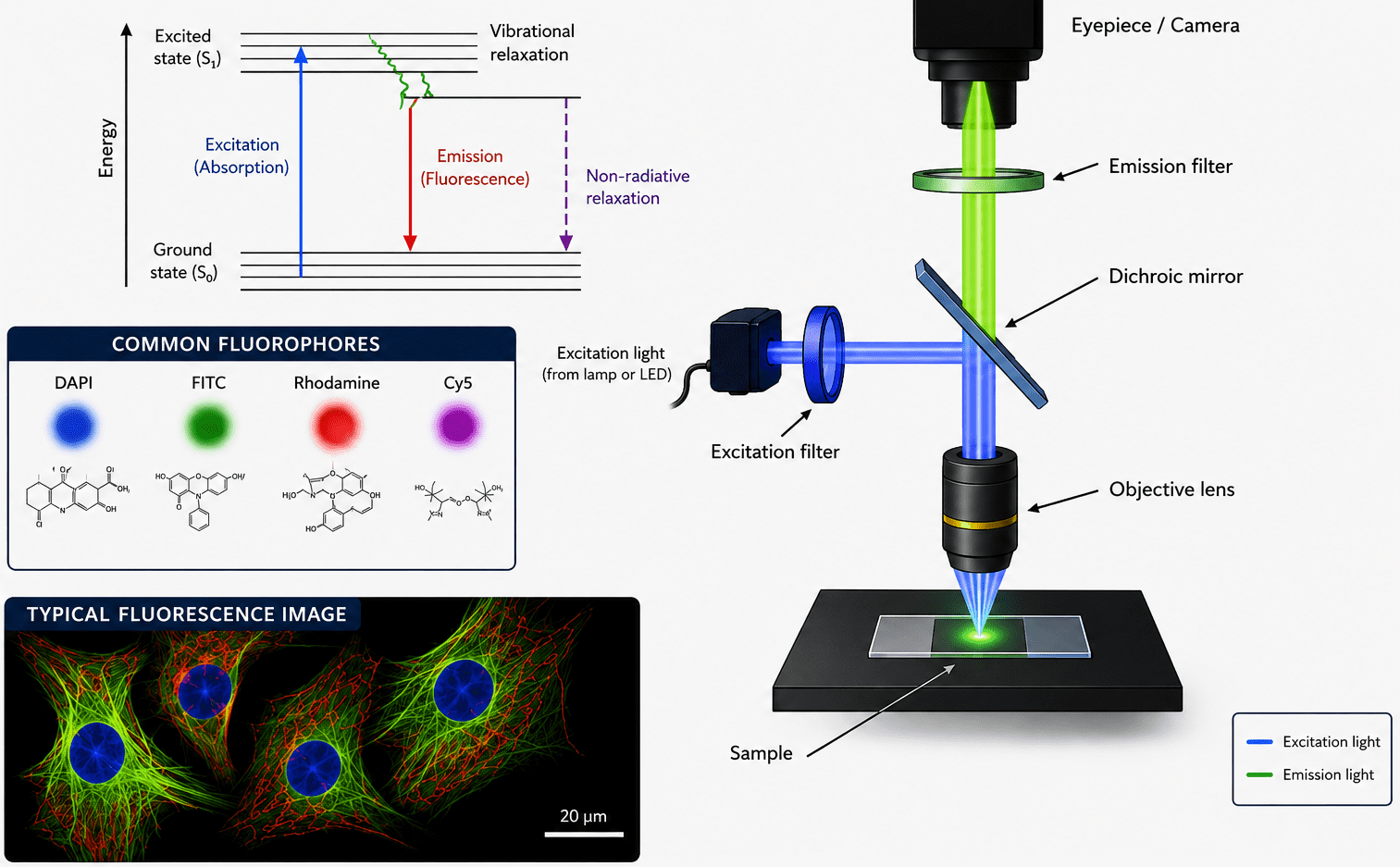
Fluorescence microscopy is an advanced optical imaging technique that utilizes the phenomenon of fluorescence to visualize cellular structures, biomolecules, and dynamic biological processes with remarkable specificity and sensitivity. By employing fluorescent molecules known as fluorophores, the technique enables selective labeling and detection of particular components within complex biological and material samples. Owing to its ability to generate high-contrast images and facilitate real-time observation of living systems, fluorescence microscopy has become an indispensable tool in diverse scientific disciplines, including cell biology, microbiology, biophysics, molecular biology, neuroscience, and medical diagnostics. Its applications range from studying intracellular organelles and protein localization to tracking microbial interactions and disease progression.
The fundamental strength of fluorescence microscopy lies in its exceptional versatility and adaptability. Unlike conventional light microscopy, which relies primarily on differences in light absorption or scattering, fluorescence microscopy provides molecular-level specificity by selectively exciting fluorescent probes and detecting their emitted light. This capability allows researchers to investigate complex biological events with high spatial and temporal resolution. Over time, the field has evolved from simple widefield fluorescence systems to sophisticated imaging platforms such as confocal microscopy, total internal reflection fluorescence (TIRF) microscopy, multiphoton microscopy, and super-resolution techniques including stimulated emission depletion microscopy (STED), photoactivated localization microscopy (PALM), and stochastic optical reconstruction microscopy (STORM). These advancements have significantly improved image resolution, sensitivity, and three-dimensional imaging capabilities.
In addition to innovations in optical instrumentation, developments in fluorophore chemistry, digital imaging technologies, and computational image analysis have further enhanced the performance and applications of fluorescence microscopy. Modern approaches increasingly incorporate artificial intelligence and machine learning algorithms for automated image processing, segmentation, and quantitative analysis, thereby enabling more accurate interpretation of complex datasets.
Although fluorescence microscopy faces certain limitations, including photobleaching, phototoxicity, autofluorescence, and diffraction-imposed resolution barriers, continuous technological progress is overcoming many of these challenges. Consequently, fluorescence microscopy remains one of the most influential and widely used imaging techniques in modern scientific research, providing unprecedented insights into cellular architecture, molecular interactions, physiological mechanisms, and pathological processes that were previously beyond the reach of conventional microscopy methods.
Fundamental principles of fluorescence
Fluorescence is a photophysical phenomenon in which certain molecules, known as fluorophores or fluorescent dyes, absorb light energy at a particular wavelength and subsequently emit light at a longer wavelength. This process occurs extremely rapidly, usually within nanoseconds, and forms the fundamental basis of fluorescence microscopy and many modern bioimaging techniques. Fluorescence enables scientists to selectively visualize specific molecules, cellular structures, or biochemical processes with high sensitivity and specificity.
When a fluorophore is illuminated with light of sufficient energy, usually ultraviolet (UV), blue, or green light, its electrons absorb this energy and become excited. The absorbed energy promotes an electron from its normal low-energy state, called the ground state (S₀), to a higher-energy excited singlet state (S₁ or higher). However, this excited state is unstable and temporary. The molecule rapidly undergoes internal energy losses before returning to the ground state by emitting light.
Importantly, the emitted light possesses lower energy and therefore a longer wavelength than the absorbed excitation light. This difference between excitation and emission wavelengths is called the Stokes shift. The Stokes shift is essential in fluorescence microscopy because it allows emitted fluorescence to be optically separated from the excitation light using filters and dichroic mirrors, enabling clear visualization of fluorescent signals.
The fluorescence process can be summarized in three major steps:
- Excitation: A photon of light strikes the fluorophore and excites an electron from the ground state (S₀) to an excited singlet state (S₁ or higher). The energy absorbed must correspond to the excitation spectrum of the fluorophore.
- Relaxation: Once excited, the electron rapidly loses a portion of its energy through non-radiative processes such as vibrational relaxation and internal conversion. During this stage, energy is dissipated as heat rather than light.
- Emission: The electron eventually returns to the ground state, releasing the remaining energy in the form of a photon. Because some energy was lost during relaxation, the emitted photon has lower energy and a longer wavelength than the excitation photon.
Fluorescence is widely exploited in biological and medical sciences because fluorophores can be attached to antibodies, nucleic acids, proteins, or cellular organelles. This selective labeling allows researchers to monitor molecular localization, interactions, and dynamic cellular events in living or fixed specimens with remarkable precision. Fluorescence microscopy illustrates the fundamental components and optical pathway involved in the generation of fluorescent images (Figure 1). Excitation light passes through an excitation filter and dichroic mirror to illuminate the specimen, where fluorophores absorb light energy and emit fluorescence at longer wavelengths. The emitted light is collected by the objective lens, filtered through an emission filter, and transmitted to the eyepiece or camera for image formation. Common fluorophores such as DAPI (4′,6-diamidino-2-phenylindole), FITC (Fluorescein Isothiocyanate), Rhodamine, and Cy5 (Cyanine 5) are widely used because they selectively label cellular structures and biomolecules with high sensitivity and specificity. Fluorescence microscopy is an essential tool in modern biological and medical research, enabling detailed visualization of cells, tissues, proteins, nucleic acids, and dynamic intracellular processes with excellent contrast and molecular precision.
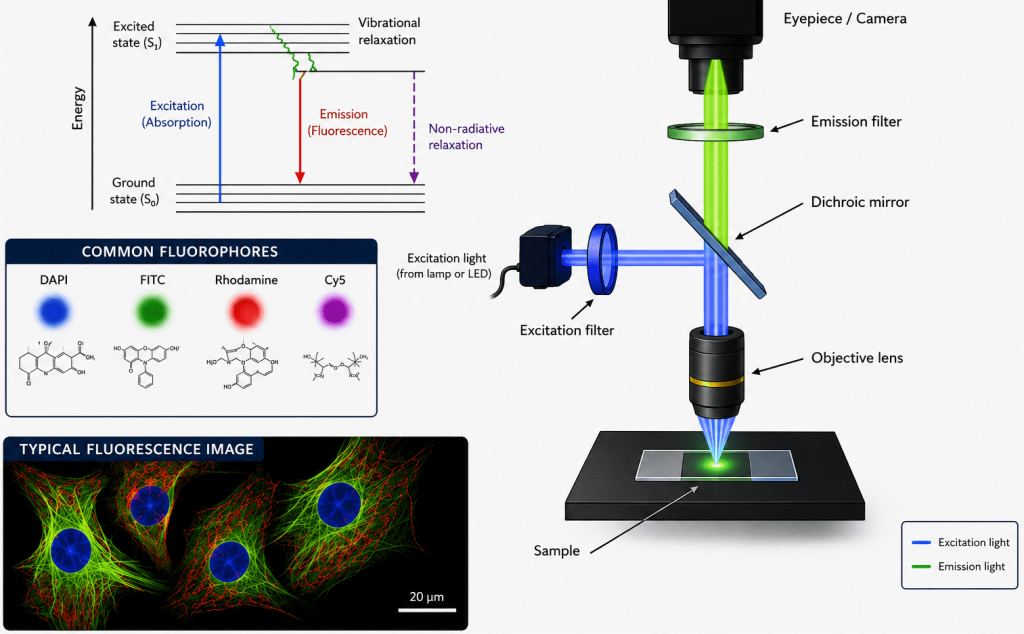
Jablonski diagram
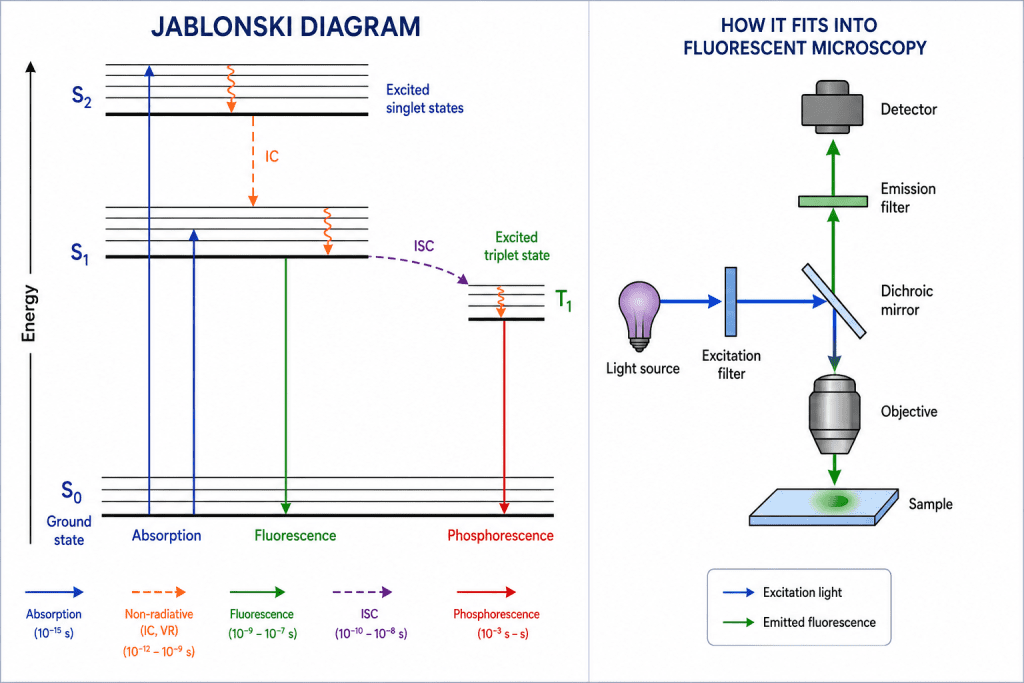
The Jablonski diagram is a graphical representation used in fluorescence spectroscopy and fluorescence microscopy to explain the electronic energy states of a molecule and the transitions that occur between them during absorption and emission of light (Figure 2). It provides a simplified visualization of how fluorophores behave when they interact with electromagnetic radiation. The diagram is named after the Polish physicist Aleksander Jablonski, who developed this model to describe photophysical processes in fluorescent molecules.
In a Jablonski diagram, the energy levels of a molecule are represented as horizontal lines. The lowest energy state is called the ground singlet state (S₀), while higher-energy excited singlet states are labeled S₁, S₂, S₃, and so on. Each electronic state contains several closely spaced vibrational energy levels, shown as multiple horizontal sublevels. These vibrational levels arise because molecules can vibrate in different ways even within the same electronic state.
When a fluorophore absorbs light, an electron is promoted from the ground state (S₀) to an excited electronic state such as S₁ or S₂. This process is represented by a vertical upward arrow and is known as excitation or absorption. The transition occurs extremely rapidly, usually within femtoseconds (10⁻¹⁵ s). Because absorption happens faster than molecular motion, the electron moves vertically to a higher vibrational level according to the Franck–Condon principle.
After excitation, the molecule is unstable and undergoes vibrational relaxation, where excess vibrational energy is lost as heat to the surrounding environment. This process brings the electron to the lowest vibrational level of the excited singlet state, usually S₁. Vibrational relaxation occurs within picoseconds and does not involve emission of light.
Another important non-radiative process shown in the Jablonski diagram is internal conversion. Internal conversion refers to the transfer of energy between electronic states of the same spin multiplicity, such as from S₂ to S₁, without emission of a photon. The energy is dissipated as molecular vibrations or heat. As a result, fluorescence emission generally occurs from the lowest excited singlet state (S₁), regardless of which higher excited state was initially populated. This observation is described by Kasha’s rule.
The most important radiative process in fluorescence microscopy is fluorescence emission. After relaxation to the lowest vibrational level of S₁, the electron returns to the ground state S₀ while emitting a photon of lower energy. This transition is represented by a downward diagonal arrow in the Jablonski diagram. Because some energy has already been lost during vibrational relaxation, the emitted photon has a longer wavelength and lower energy than the absorbed photon. This difference between excitation and emission wavelengths is called the Stokes shift, a fundamental property that allows fluorescence signals to be separated from excitation light using optical filters.
The diagram also includes intersystem crossing (ISC), a process in which the excited electron undergoes a spin conversion from an excited singlet state (S₁) to an excited triplet state (T₁). Triplet states have electrons with parallel spins and are generally lower in energy and longer-lived than singlet states. Intersystem crossing is a non-radiative transition and is particularly common in molecules containing heavy atoms or certain chemical structures that facilitate spin-orbit coupling.
Once the molecule enters the triplet state, it may return to the ground state through a slow radiative process known as phosphorescence. In the Jablonski diagram, phosphorescence is shown as a downward arrow from T₁ to S₀. Because this transition involves a change in electron spin, it is quantum mechanically forbidden and therefore occurs much more slowly than fluorescence. Fluorescence lifetimes are typically in the nanosecond range, whereas phosphorescence can persist from milliseconds to several seconds or even longer.
The Jablonski diagram is essential for understanding several practical phenomena in fluorescence microscopy, including fluorescence lifetime, photobleaching, energy transfer, and quantum yield. It explains why fluorophores emit light at longer wavelengths, why some dyes bleach rapidly, and how different relaxation pathways compete with fluorescence emission. Consequently, the Jablonski diagram serves as the theoretical foundation for advanced fluorescence techniques such as Förster Resonance Energy Transfer (also called Fluorescence Resonance Energy Transfer – FRET), FLIM (fluorescence lifetime imaging microscopy), confocal microscopy, and super-resolution imaging.
Key fluorescence parameters of fluorescence microscopy
Fluorescence microscopy performance and interpretability depend fundamentally on a small set of photophysical parameters that govern how fluorophores absorb, store, and re-emit energy. These parameters (which include quantum yield, fluorescence lifetime, photobleaching, and brightness) determine signal quality, quantitative reliability, and imaging sensitivity in both fixed and live systems. The most critical include quantum yield, fluorescence lifetime, photobleaching kinetics, and molecular brightness. Each parameter reflects a different aspect of the excited-state dynamics of fluorophores and collectively defines how “useful” a fluorescent probe is in microscopy applications.
1. Quantum yield (φ): efficiency of photon conversion
Quantum yield (Φ) is a dimensionless parameter that quantifies the efficiency with which an excited fluorophore converts absorbed photons into emitted fluorescence photons. It is formally defined as:
Φ = Number of photons emitted / Number of photons absorbed
Physical interpretation of quantum yield
After absorbing a photon, a fluorophore enters an excited electronic state. From this state, it can relax via:
- Radiative decay (fluorescence emission)
- Non-radiative decay (heat dissipation, vibrational relaxation, internal conversion)
Quantum yield reflects the competition between these pathways. A high Φ indicates that radiative decay dominates, whereas a low Φ indicates that energy is predominantly lost non-radiatively.
Typical values of quantum yield
- Organic dyes: ~0.1 to 1.0
- Fluorescent proteins: ~0.2 to 0.8
- Quantum dots: often >0.5 and can approach unity
Importance of quantum yield in microscopy
- Direct determinant of fluorescence signal strength
- Critical for weak-signal applications (e.g., single-molecule imaging)
- Influences exposure time and laser power requirements
Key limitation of quantum yield
Quantum yield is environment-dependent; it can change with:
- pH
- Polarity
- Ion concentration
- Protein binding or conformational state
Thus, Φ is not always a fixed molecular constant in biological systems.
2. Fluorescence lifetime: excited-state decay kinetics
Fluorescence lifetime (τ) is the average time a fluorophore remains in its excited state before returning to the ground state via photon emission. It is typically on the order of nanoseconds (10⁻⁹ s).
Formal definition of fluorescence lifetime
r = 1 / kr + knr
Where:
- kr = radiative decay rate constant
- knr= non-radiative decay rate constant
Key characteristics of fluorescence lifetime
- Independent of fluorophore concentration
- Sensitive to local microenvironment
- Measured using time-resolved techniques (e.g., FLIM)
Biological sensitivity of fluorescence lifetime
Fluorescence lifetime changes in response to:
- Protein binding interactions
- Ionic strength and viscosity
- Oxygen concentration
- pH variations
- Förster resonance energy transfer (FRET)
Applications of fluorescence lifetime in microscopy
- Fluorescence lifetime imaging microscopy (FLIM) enables spatial mapping of τ across a sample
- Distinguishes fluorophores with overlapping emission spectra
- Quantifies molecular interactions (especially FRET efficiency)
Analytical significance of fluorescence lifetime
Unlike intensity-based measurements, lifetime is largely independent of:
- fluorophore concentration
- excitation intensity
- light path fluctuations
This makes it a robust parameter for quantitative imaging.
3. Photobleaching: irreversible fluorescence loss
Photobleaching refers to the irreversible destruction of a fluorophore’s ability to fluoresce due to photochemical reactions induced by prolonged illumination.
Mechanistic basis of photobleaching
Photobleaching arises when excited fluorophores:
- React with molecular oxygen to form reactive oxygen species (ROS)
- Undergo bond cleavage or structural rearrangement
- Enter long-lived triplet states that promote chemical degradation
Kinetic behavior of photobleaching
Photobleaching is often modeled as an exponential decay:
I(t) = I0e-kt
Where:
- I(t) = fluorescence intensity at time t
- k = bleaching rate constant
- I0= initial intensity
Contributing factors of photobleaching
- Excitation intensity (higher power → faster bleaching)
- Oxygen concentration
- Environmental antioxidants or antifade reagents
- Fluorophore chemical stability
- Imaging modality (widefield vs confocal vs two-photon)
Experimental consequences of photobleaching
- Loss of signal over time in time-lapse imaging
- Reduced accuracy in quantitative intensity measurements
- Limitation in long-duration live-cell imaging
Mitigation strategies of photobleaching
- Use of oxygen scavenging systems
- Low-light imaging strategies
- More photostable fluorophores (e.g., quantum dots)
- Anti-fade mounting media
Photobleaching is not merely a technical inconvenience; it fundamentally constrains temporal resolution and observation window in fluorescence experiments.
4. Brightness: overall photon output efficiency
Brightness is a composite parameter that defines how intense a fluorophore appears under given excitation conditions. It is generally expressed as:
Brightness = ε . Φ
Where:
- ε = molar extinction coefficient (light absorption efficiency)
- Φ = quantum yield
Interpretation of brightness
Brightness integrates:
- How strongly a molecule absorbs excitation light (ε)
- How efficiently it emits fluorescence (Φ)
Thus, a fluorophore can be:
- Highly absorbing but weakly emitting → moderate brightness
- Weakly absorbing but highly efficient → moderate brightness
- High in both → extremely bright probe
Practical significance of brightness
Brightness determines:
- Signal-to-noise ratio in images
- Required laser power or exposure time
- Detectability of low-abundance targets
- Performance in single-molecule detection
Comparative behavior of brightness
- Organic dyes: moderate to high brightness
- Fluorescent proteins: moderate brightness (often lower ε)
- Quantum dots: extremely high brightness due to large absorption cross-section
Imaging implications of brightness
- Bright probes reduce phototoxicity (less excitation needed)
- Enable faster imaging rates
- Improve deep-tissue imaging feasibility
5. Interdependence of fluorescence parameters
Although often treated separately, these parameters are physically interconnected:
- Quantum yield and lifetime share dependence on radiative vs non-radiative decay rates
- Brightness depends directly on quantum yield
- Photobleaching competes with fluorescence emission pathways and can reduce both apparent quantum yield and lifetime
- Environmental conditions can simultaneously alter all four parameters
This coupling means fluorophore performance cannot be fully understood from a single metric; instead, a multidimensional photophysical profile is required. Together, these parameters define whether a fluorophore is suitable for:
- fixed-cell imaging
- live-cell dynamics
- super-resolution techniques
- single-molecule detection
- long-term time-lapse experiments
Components of a fluorescence microscope
A fluorescence microscope differs fundamentally from a standard bright-field light microscope not only in how it illuminates a specimen but also in how it selectively detects light emitted from fluorescent molecules. Instead of simply transmitting light through a sample, it relies on a carefully engineered optical pathway designed to separate weak fluorescence emission from much stronger excitation light. This is achieved through a combination of specialized components that work in sequence: the light source, excitation filter, dichroic mirror, emission filter, objective lens, and detector.
In fluorescence microscopy, each component plays a specialized role in ensuring that weak fluorescent signals can be efficiently excited, isolated, and detected with high fidelity. The light source provides controlled excitation energy, the excitation filter ensures spectral specificity, the dichroic mirror directs light appropriately, the emission filter isolates the fluorescent signal, the objective lens collects and focuses emitted photons, and the detector converts them into a usable image. Together, these components form a highly optimized optical system that enables sensitive, high-resolution visualization of molecular and cellular processes.
The major components of fluorescence microscope include:
- Light source
- Excitation filter
- Dichroic mirror (beam splitter)
- Emission Filter
- Objective lens
- Detector
Light source
The light source is the origin of excitation energy in fluorescence microscopy. Because fluorescence requires electrons in fluorophores to be raised to higher energy states, the illumination must be sufficiently intense and spectrally appropriate to match the absorption properties of the fluorophore.
Several types of light sources are used depending on the application:
- Mercury arc lamps: These were historically the most common light source. They produce very intense light with strong discrete emission lines, particularly in the UV and blue regions. This makes them suitable for exciting many classical fluorophores. However, they have limitations such as instability over time, high heat generation, and relatively short lifespan.
- Xenon lamps: These provide a more continuous and uniform spectrum across the visible range compared to mercury lamps. This makes them more versatile for multi-fluorophore experiments. They are particularly useful when broad wavelength coverage is required, although they are less intense at specific peaks than mercury lamps.
- LEDs (light emitting diodes): Modern fluorescence microscopes increasingly use LEDs because they are stable, energy-efficient, long-lasting, and capable of rapid switching between wavelengths. LEDs also produce less heat, reducing sample damage during live imaging. Their spectral output is narrower, which is advantageous for precise excitation control.
- Lasers: In advanced fluorescence systems such as confocal, multiphoton, and super-resolution microscopes, lasers are the primary excitation source. They provide highly coherent, monochromatic, and intense light, allowing precise spatial and temporal control. Different laser lines correspond to specific excitation wavelengths for targeted fluorophores.
Excitation filter
The excitation filter is responsible for selecting only the wavelengths of light that will effectively excite the fluorophore of interest. Since most light sources emit a broad spectrum, this filter is essential for ensuring specificity.
It works by:
- Allowing a narrow band of wavelengths to pass through (band-pass filter), or
- Blocking unwanted wavelengths while transmitting desired excitation light.
By restricting excitation to a defined spectral range, the excitation filter minimizes background noise and reduces unwanted stimulation of other fluorophores in multi-label experiments.
Dichroic mirror (beam splitter)
The dichroic mirror is one of the most critical optical components in fluorescence microscopy. It is positioned at a 45-degree angle in the light path and has wavelength-selective reflective and transmissive properties.
The function of beam splitter is dual:
- Reflection of excitation light: The dichroic mirror reflects the selected excitation wavelengths down through the objective lens toward the specimen.
- Transmission of emission light: Once the fluorophore emits light at a longer wavelength, the dichroic mirror allows this emission to pass upward toward the detector.
This selective behavior is based on thin-film interference coatings that are engineered to discriminate between excitation and emission wavelengths, ensuring maximum signal separation and efficiency.
Emission filter
After fluorescence emission is generated by the sample, it still contains potential contamination from reflected excitation light or scattered photons. The emission filter ensures that only the desired fluorescent signal reaches the detector.
Key functions include:
- Blocking residual excitation light that may have passed through the system
- Selecting a narrow emission wavelength range specific to the fluorophore
This improves contrast and significantly enhances signal-to-noise ratio, which is critical when detecting weak fluorescent signals in biological samples.
Objective lens
The objective lens is central to both excitation delivery and emission collection. In fluorescence microscopy, its importance is amplified because fluorescence signals are typically weak and require efficient light gathering.
Key characteristics include:
- High numerical aperture (NA): A high NA (e.g., 1.3-1.49 in oil immersion lenses) is essential for maximizing both resolution and photon collection efficiency. The NA determines how much emitted light can be captured by the lens.
- Resolution control: The objective largely determines the optical resolution of the system, influencing the ability to distinguish fine structural details.
- Brightness and sensitivity: Higher NA objectives capture more emitted photons, increasing image brightness.
- Depth of field: Fluorescence microscopy often uses thin optical sections; the objective helps define how much of the sample thickness is in focus.
Oil, water, or glycerol immersion objectives are frequently used to match refractive indices and reduce light scattering.
Detector
The detector converts emitted photons into a measurable signal, ultimately producing the fluorescence image. Different detector types are used depending on whether imaging is visual, digital, or quantitative.
- Eyepiece (ocular lens): In basic systems, the fluorescence image can be viewed directly by the observer. However, this method is limited in sensitivity and quantitative capability.
- Charge-Coupled Device (CCD) / Complementary Metal-Oxide-Semiconductor (CMOS) cameras: These are widely used in modern fluorescence microscopy for digital imaging. They provide high sensitivity, spatial resolution, and the ability to capture time-lapse sequences. CMOS cameras are particularly popular due to fast readout speeds and low noise.
- Photomultiplier tubes (PMTs): Used mainly in scanning systems such as confocal microscopy. PMTs are extremely sensitive detectors capable of detecting very low light levels by amplifying photon signals through electron multiplication. This makes them ideal for imaging dim fluorescent samples or thin optical sections.
Fluorophores and labeling strategies
Fluorophores are the core functional units in fluorescence microscopy because they translate molecular and structural information into detectable optical signals. A fluorophore is a molecule that absorbs light at a defined excitation wavelength and emits light at a longer emission wavelength, a shift known as the Stokes shift. The selection of fluorophores and labeling strategies critically determines experimental performance, influencing specificity, sensitivity, photostability, and suitability for live or fixed imaging. As a result, careful fluorophore choice is fundamental to robust experimental design.
Fluorophores fall into several major classes. Organic dyes such as fluorescein and rhodamine are widely used due to their high brightness and chemical versatility. Fluorescent proteins, including green fluorescent proteins (GFP) variants, enable genetically encoded labeling, making them especially powerful for live-cell imaging and dynamic studies. Quantum dots, by contrast, are nanocrystal-based fluorophores characterized by exceptional photostability, broad excitation spectra, and narrow emission peaks, which make them ideal for multiplexing applications.
These fluorophores are introduced into biological systems using complementary labeling strategies. Immunofluorescence relies on antibodies for highly specific target recognition, genetic tagging fuses fluorescent proteins to proteins of interest for in vivo expression, and chemical stains bind directly to cellular components such as nucleic acids or membranes. Multiplexing combines multiple fluorophores simultaneously to resolve complex molecular architectures, though it requires careful spectral separation and calibration to avoid signal overlap. Together, these approaches enable fluorescence microscopy to achieve high molecular specificity while preserving spatial and temporal resolution in biological systems.
Types of fluorophores
Organic dyes
Organic fluorescent dyes are small synthetic molecules that have been the backbone of fluorescence microscopy for decades.
Examples of organic dye include:
- Fluorescein
- Rhodamine
- DAPI
Advantages of organic dyes
Organic dyes are widely used because they are:
- Highly bright: They have strong absorption coefficients and efficient fluorescence emission.
- Chemically versatile: They can be conjugated to antibodies, nucleic acids, or small ligands.
- Spectrally diverse: Available across UV, visible, and near-infrared ranges.
Limitations of organic dyes
Organic dyes also have important limitation. Despite these limitations, organic dyes remain indispensable for fixed-sample imaging and high-specificity labeling protocols. The limitations of organic dyes include:
- Photobleaching susceptibility: Organic dyes degrade under prolonged excitation, especially under high-intensity illumination.
- Environmental sensitivity: Their fluorescence can vary with pH, polarity, or local chemical conditions.
- Limited photostability in live imaging: This restricts their use in long-term dynamic studies.
Fluorescent proteins
Fluorescent proteins revolutionized live-cell imaging by enabling genetically encoded fluorescence. The most iconic example of a fluorescent protein is: Green Fluorescent Protein (GFP). GFP is a 26.9 kDa fluorescent protein that emits bright green fluorescence (~509 nm) upon excitation with blue or ultraviolet light. Owing to its intrinsic fluorescence, low cytotoxicity, and genetic encodability, GFP has become one of the most widely used molecular markers in cell and molecular biology for monitoring gene expression, protein localization, intracellular trafficking, and dynamic cellular processes in living cells.
GFP has some major benefits. Fluorescent proteins are:
- Genetically encoded: The gene encoding the fluorescent protein is fused to a gene of interest.
- Self-synthesizing chromophores: They form their fluorescent structure through internal post-translational chemistry.
- Ideal for live-cell imaging: They allow continuous observation of protein localization and dynamics in living systems without external staining.
Advantages of GFP:
- Minimal sample perturbation
- High specificity via genetic fusion
- Stable expression over time
Limitations of GFP:
- Larger size compared to small dyes, which may interfere with protein function
- Maturation time required before fluorescence appears
- Generally lower brightness compared to some synthetic dyes
Despite these trade-offs, fluorescent proteins are foundational in molecular and cell biology, enabling real-time tracking of gene expression, protein trafficking, and cellular dynamics.
Quantum dots
Quantum dots are inorganic semiconductor nanocrystals with unique photophysical properties.
They exhibit:
- Extremely high brightness
- Superior photostability (resistant to photobleaching)
- Size-tunable emission spectra, meaning emission wavelength depends on particle size rather than chemical structure
- Narrow emission peaks with broad excitation ranges
These properties of quantum dots allow:
- Simultaneous excitation of multiple quantum dots using a single light source
- Highly multiplexed imaging with minimal spectral overlap
However, they also present some challenges such as:
- Potential cytotoxicity (especially heavy metal-based formulations)
- Larger physical size compared to organic dyes
- Complex surface chemistry required for biological conjugation
Quantum dots are particularly valuable in long-term imaging and single-particle tracking applications.
Labeling techniques in fluorescence microscopy
The utility of fluorophores depends heavily on how they are attached to biological targets. Several labeling strategies are commonly used in fluorescence microscopy as follows:
Immunofluorescence in fluorescence microscopy
Immunofluorescence relies on antibodies conjugated to fluorophores to detect specific antigens (proteins, peptides, or cellular structures).
There are two main types of immunofluorescences:
- Direct immunofluorescence: Primary antibody is directly labeled with a fluorophore.
- Indirect immunofluorescence: A labeled secondary antibody binds to an unlabeled primary antibody.
Advantages of immunofluorescence:
- High specificity due to antigen–antibody recognition
- Signal amplification in indirect methods
- Wide applicability in fixed-cell and tissue imaging
Limitations of immunofluorescence:
- Requires fixation and permeabilization (not always compatible with live imaging)
- Potential for nonspecific binding or cross-reactivity
Genetic tagging in fluorescence microscopy
Genetic tagging involves fusion of fluorescent protein genes to genes of interest, producing chimeric proteins.
A classic example of this scenario is:
- Green Fluorescent Protein (GFP) fusion constructs
Advantages of genetic tagging:
- Enables real-time tracking of proteins in living cells
- High spatial and temporal resolution
- No need for external staining
Limitations of genetic tagging:
- Genetic modification required
- Potential interference with protein folding, localization, or function
- Requires validation of biological integrity of fusion constructs
Chemical stains in fluorescence microscopy
Chemical stains are small molecules that selectively bind to specific cellular structures.
Examples of chemical stains using in fluorescence microscopy include:
- DNA-binding dyes (e.g., nuclear stains like DAPI)
- Membrane dyes
- Cytoskeletal stains
Advantages of chemical stains:
- Simple and fast application
- High affinity for target structures
- Suitable for both fixed and sometimes live cells
Limitations of chemical stains:
- Lower specificity compared to antibodies or genetic tags
- Possible toxicity in live cells
- Limited ability to distinguish closely related molecular targets
Multiplexing in fluorescence microscopy
Multiplexing refers to the simultaneous imaging of multiple targets in a single sample using different fluorophores. This is achieved by selecting fluorophores with:
- Distinct excitation spectra
- Well-separated emission spectra
By combining multiple fluorescent labels, researchers can:
- Visualize different proteins within the same cell
- Study molecular interactions and spatial organization
- Map complex biological systems such as tissues or microbial communities
Key challenges in multiplexing include:
- Spectral overlap (emission bleed-through between channels)
- Requirement for careful filter selection and calibration
- Increased computational complexity in image separation and quantification
Advanced systems use spectral unmixing algorithms and highly engineered fluorophores to overcome these limitations, enabling highly multiplexed imaging of cellular processes.
Types of fluorescence microscopy
There are different types of fluorescence microscopes that exist (Figure 3). Fluorescence microscopy has evolved into several specialized forms, each designed to overcome particular imaging challenges and to provide specific types of information about biological or material samples. These techniques differ mainly in illumination methods, detection systems, resolution, imaging depth, and applications. The following are the different type of fluorescence microscopy:
Widefield fluorescence microscopy
Widefield fluorescence microscopy is the simplest and most commonly used form of fluorescence imaging. In this technique, the entire specimen is illuminated simultaneously using a broad light source such as a mercury lamp, light-emitting diode (LED), or xenon lamp. The emitted fluorescence from all regions of the sample is collected by the objective lens and detected by a camera or eyepiece.
This method is particularly suitable for thin specimens because minimal out-of-focus light is produced in thin samples. It allows rapid image acquisition, making it useful for routine laboratory observations and time-lapse imaging. The setup is relatively inexpensive and easy to operate compared to more advanced fluorescence systems.
However, one major limitation is that fluorescence emitted from out-of-focus planes is also collected. This produces blurred images and reduced contrast, especially in thick specimens. As a result, widefield microscopy is less effective for detailed three-dimensional imaging of complex tissues.
Confocal laser scanning microscopy (CLSM)
Confocal laser scanning microscopy was developed to overcome the problem of out-of-focus blur. In CLSM, a highly focused laser beam scans the sample point by point. The emitted fluorescence passes through a small pinhole aperture before reaching the detector. This pinhole blocks out-of-focus light, allowing only fluorescence from the focal plane to be detected.
Because of this optical sectioning capability, confocal microscopy produces sharp, high-contrast images and enables the reconstruction of three-dimensional structures from multiple optical sections. It is widely used in cell biology, microbiology, neuroscience, and pathology.
Confocal microscopy provides significantly improved spatial resolution compared to widefield microscopy. It is particularly valuable for imaging thick biological tissues and intracellular structures. However, the scanning process can be relatively slow, especially for large samples. The intense laser illumination may also cause photobleaching and phototoxicity, potentially damaging living cells during prolonged imaging.

Spinning disk confocal microscopy
Spinning disk confocal microscopy is a modified form of confocal imaging designed for faster image acquisition. Instead of scanning a single laser beam across the sample, this technique uses a rotating disk containing multiple pinholes. These pinholes scan many points simultaneously, greatly increasing imaging speed.
This method is especially useful for live-cell imaging because it reduces photobleaching and phototoxicity while allowing rapid capture of dynamic cellular processes. Researchers commonly use spinning disk systems to study vesicle transport, cytoskeletal movement, and intracellular signaling in real time. Although spinning disk microscopy is faster than conventional confocal microscopy, it may provide slightly lower optical sectioning quality in very thick specimens.
Total internal reflection fluorescence (TIRF) microscopy
Total Internal Reflection Fluorescence microscopy is a specialized technique used to study events occurring very close to the cell surface. In TIRF microscopy, light undergoes total internal reflection at the interface between glass and the specimen. This creates an evanescent wave that excites fluorophores only within approximately 100-200 nm of the surface.
Because fluorescence excitation is restricted to a very thin region, background fluorescence is dramatically reduced, producing extremely high contrast images. TIRF microscopy is widely used to study plasma membrane dynamics, receptor trafficking, exocytosis, endocytosis, and single-molecule interactions. The main limitation of TIRF microscopy is that it can only image structures near the surface of the sample and cannot visualize deeper cellular regions.
Two-photon microscopy
Two-photon microscopy is an advanced fluorescence imaging technique that uses the simultaneous absorption of two low-energy infrared photons to excite fluorophores. Since excitation occurs only at the focal point where photon density is highest, photodamage outside the focal region is minimized.
Infrared light penetrates deeper into biological tissues than visible light, allowing imaging several hundred micrometers below the surface. This makes two-photon microscopy particularly valuable for neuroscience, developmental biology, and in vivo imaging.
Advantages include reduced phototoxicity, decreased photobleaching, and deeper tissue penetration. However, the technique requires expensive pulsed lasers and sophisticated optical systems, making it more costly and technically demanding.
Super-resolution microscopy
Traditional fluorescence microscopy is limited by optical diffraction to a resolution of approximately 200 nm. Super-resolution microscopy techniques overcome this barrier and achieve nanometer-scale resolution.
Major super-resolution methods include:
- STED (Stimulated Emission Depletion)
- PALM (Photoactivated Localization Microscopy)
- STORM (Stochastic Optical Reconstruction Microscopy)
These techniques allow visualization of cellular structures and molecular arrangements at resolutions as low as 10-50 nm. Super-resolution microscopy has revolutionized cell biology by enabling detailed imaging of protein complexes, cytoskeletal organization, and membrane nanostructures. Despite its extraordinary resolution, super-resolution microscopy often requires specialized fluorophores, complex instrumentation, and extensive image processing.
Fluorescence lifetime imaging microscopy (FLIM)
Fluorescence Lifetime Imaging Microscopy measures the fluorescence decay time of fluorophores rather than fluorescence intensity. Fluorescence lifetime is highly sensitive to the local chemical environment, including pH, ion concentration, oxygen levels, and molecular interactions. FLIM is particularly useful for studying cellular metabolism, molecular binding events, and intracellular signaling pathways. Since fluorescence lifetime is independent of fluorophore concentration, FLIM can provide more reliable quantitative data than intensity-based methods.
Förster resonance energy transfer (FRET)
Förster Resonance Energy Transfer is a fluorescence-based technique used to detect molecular interactions at extremely short distances. In FRET, energy is transferred from an excited donor fluorophore to a nearby acceptor fluorophore without emission of a photon. FRET occurs only when the two fluorophores are within approximately 1-10 nm of each other, making it an effective molecular ruler. It is widely used to study protein-protein interactions, conformational changes, receptor activation, and intracellular signaling mechanisms. This technique provides valuable information about molecular proximity and dynamics that cannot be obtained using conventional fluorescence microscopy alone.
Resolution and optical considerations in fluorescence microscopy
Resolution is one of the most important parameters in fluorescence microscopy because it determines the ability to distinguish two closely spaced objects as separate entities. The quality of fluorescence images depends not only on the fluorescent labels used but also on several optical factors, including diffraction, numerical aperture, wavelength of light, and signal-to-noise ratio.
Diffraction limit
The resolution of conventional fluorescence microscopes is fundamentally limited by the diffraction of light. This concept was described by Ernst Abbe and is represented by Abbe’s equation:
d = λ / 2NA
where:
- d = minimum resolvable distance,
- λ = wavelength of emitted light,
- NA = numerical aperture of the objective lens.
According to this equation, resolution improves when shorter wavelengths and higher numerical aperture lenses are used. In standard fluorescence microscopy, the lateral (x-y) resolution is typically about 200 nm, while axial (z-axis) resolution is around 500-700 nm. As a result, structures closer than these distances may appear merged together. The diffraction barrier was historically a major limitation until the development of super-resolution microscopy techniques such as STED, PALM, and STORM, which can achieve nanometer-scale resolution.
Numerical aperture (NA)
The numerical aperture of the objective lens is another critical optical parameter. It determines the light-gathering capacity and resolving power of the microscope. Numerical aperture depends on the refractive index of the medium between the specimen and objective lens, as well as the angle at which light enters the lens.
Objectives with higher NA values collect more emitted fluorescence and provide brighter, sharper images. Oil immersion objectives often have NA values of 1.3-1.4, significantly improving image resolution compared to dry objectives. High NA lenses are especially important in fluorescence microscopy because fluorescence signals are often weak and require efficient light collection. However, higher NA lenses generally have a shallower depth of field and shorter working distance.
Signal-to-noise ratio (SNR)
The signal-to-noise ratio (SNR) is essential for producing high-quality fluorescence images. The “signal” refers to fluorescence emitted from the labeled target, while “noise” includes unwanted background signals such as autofluorescence, detector noise, and scattered light. A high SNR produces clear and detailed images, whereas a low SNR results in poor contrast and difficulty distinguishing structures.
Several factors influence SNR, including:
- Detector sensitivity,
- Fluorophore brightness,
- Exposure time,
- Background fluorescence,
- Optical alignment,
- Sample preparation quality.
Modern detectors such as CCD and CMOS cameras improve SNR through enhanced sensitivity and reduced electronic noise.
Sample preparation in fluorescence microscopy
Proper sample preparation is essential for obtaining reliable fluorescence microscopy results. Poor preparation can lead to weak fluorescence, structural distortion, or high background noise.
Fixation: Fixation preserves cellular and tissue structures by stabilizing proteins and membranes. It prevents degradation and maintains morphology during staining and imaging.
Chemical fixation: Common chemical fixatives include:
- Formaldehyde: Preserves protein structure while maintaining antigenicity.
- Glutaraldehyde: Provides stronger cross-linking but may increase autofluorescence.
Cryofixation: Cryofixation rapidly freezes samples, preserving structures in a near-native state. It minimizes artifacts caused by chemical treatments and is useful for high-resolution imaging.
Permeabilization: Permeabilization allows fluorescent dyes or antibodies to penetrate cell membranes and access intracellular targets. Detergents such as Triton X-100 and saponin are commonly used. Excessive permeabilization can damage cellular structures, so conditions must be carefully optimized.
Mounting media: Mounting media preserve stained samples and improve optical clarity. They help maintain the refractive index between the specimen and objective lens while reducing photobleaching. Many mounting solutions contain antifade reagents that protect fluorophores from oxidative damage during illumination.
Live-cell imaging: Live-cell fluorescence microscopy enables observation of dynamic biological processes in real time. Successful live imaging requires:
- Non-toxic fluorescent probes,
- Controlled temperature and CO2 levels,
- Minimal phototoxicity and photobleaching.
Special environmental chambers are often used to maintain physiological conditions throughout imaging experiments.
Advantages of fluorescence microscopy
Fluorescence microscopy offers several major advantages that make it one of the most widely used imaging techniques in biological and medical research. One of its greatest strengths is its high specificity, as fluorescent dyes or labeled antibodies can bind selectively to particular molecules, proteins, organelles, or nucleic acids within a sample. This enables researchers to visualize precise cellular components without interference from surrounding structures. Another important advantage is its high sensitivity. Fluorescence microscopy can detect extremely small amounts of biological material, and advanced systems are even capable of detecting single molecules, making it highly valuable in molecular biology and diagnostic applications.
The technique also possesses strong multiplexing capability, meaning multiple fluorophores with different emission wavelengths can be used simultaneously in the same specimen. This allows several targets to be observed at once, helping researchers study complex cellular interactions and pathways. In addition, fluorescence microscopy supports live-cell imaging, enabling real-time observation of dynamic biological processes such as cell division, intracellular transport, and microbial interactions without destroying the sample. Furthermore, advanced fluorescence systems such as confocal microscopy provide excellent three-dimensional (3D) imaging capabilities by collecting optical sections at different depths. These sections can be reconstructed into detailed 3D representations of tissues or cells, improving structural analysis and spatial understanding.
Limitations and challenges
Fluorescence microscopy, despite its high sensitivity and specificity, has several important limitations and technical challenges that can affect image quality and experimental accuracy. One major issue with fluorescence microscopy is photobleaching. Photobleaching refers to the gradual and irreversible loss of fluorescence as fluorophores are repeatedly exposed to excitation light. This reduces signal intensity over time and can limit long-term imaging experiments. Photobleaching can be minimized by using lower light intensity, reducing exposure time, and applying antifade reagents in the mounting medium.
Another significant challenge is phototoxicity, particularly during live-cell imaging. Intense illumination, especially ultraviolet (UV) light, can damage cellular structures, alter physiological processes, and even lead to cell death. This makes careful optimization of imaging conditions essential. Autofluorescence is also problematic in many biological samples. Certain cellular molecules and tissues naturally emit fluorescence, creating background signals that interfere with the detection of labeled targets and reduce image contrast and sensitivity.
A further limitation is spectral overlap, where the emission spectra of different fluorophores overlap with one another. This can cause signal bleed-through between imaging channels and complicate multicolor experiments. Proper fluorophore selection and filter optimization are necessary to reduce this issue. Conventional fluorescence microscopy is restricted by the diffraction limit of light, which limits resolution to approximately 200 nm. Although super-resolution techniques can overcome this barrier, they often require advanced instrumentation and complex image analysis.
Applications of fluorescence microscopy
Fluorescence microscopy has extensive applications in biological, medical, and material sciences because it enables highly specific visualization of structures and molecules within complex samples. In cell biology, it is widely used for organelle visualization, allowing researchers to study structures such as the nucleus, mitochondria, lysosomes, and endoplasmic reticulum in living or fixed cells. It also plays a major role in analyzing cytoskeleton organization by staining microtubules, actin filaments, and intermediate filaments, which helps in understanding cell movement, division, and structural stability. Protein localization studies using fluorescent tags such as GFP provide insights into protein trafficking, interactions, and cellular functions.
In molecular biology, fluorescence microscopy is important for monitoring gene expression and intracellular signaling pathways. Fluorescent probes and reporter genes allow researchers to track when and where genes are activated. RNA localization studies help determine the spatial distribution of messenger RNA within cells, providing information about gene regulation and protein synthesis.
In microbiology, fluorescence microscopy is used for the identification and differentiation of microorganisms, including bacteria, fungi, and viruses. Fluorescent stains and probes enable rapid detection of pathogens and analysis of microbial communities. It is also essential in biofilm research, where fluorescent markers reveal the architecture and viability of microbial populations. Additionally, fluorescence techniques are widely used in antibiotic resistance studies to track resistant bacterial strains and examine the spread of antimicrobial resistance genes.
In medical diagnostics, fluorescence microscopy contributes to cancer detection, immunohistochemistry, and pathogen identification. In neuroscience, it is used for neural circuit mapping and calcium imaging to study neuronal activity. In materials science, fluorescence microscopy assists in polymer characterization and visualization of nanomaterials, helping researchers analyze material composition, structure, and interactions at microscopic levels.
References
Jeff W. Lichtman, & José-Angel Conchello. (2005). Fluorescence microscopy. Nature Methods, 2(12), 910–919.
Roger Y. Tsien. (1998). The green fluorescent protein. Annual Review of Biochemistry, 67, 509–544.
Bernard Valeur. (2002). Molecular fluorescence: Principles and applications. Wiley-VCH.
Joseph R. Lakowicz. (Ed.). (1999). Principles of fluorescence spectroscopy (2nd ed.). Plenum Press.
Michael J. Sanderson, Ian Smith, Ian Parker, & Martin D. Bootman. (2014). Fluorescence microscopy. Cold Spring Harbor Protocols, 2014(10), pdb.top071795.
Ulrich Kubitscheck (Ed.). (2017). Fluorescence microscopy: From principles to biological applications. Wiley-VCH.
S. C. M. Reinhardt, Luciano A. Masullo, Isabelle Baudrexel, Philipp R. Steen, Rafal Kowalewski, Alexandra S. Eklund, Sebastian Strauss, Eduard M. Unterauer, Thomas Schlichthaerle, Maximilian T. Strauss, Christian Klein, & Ralf Jungmann. (2023). Ångström-resolution fluorescence microscopy. Nature, 617(7960), 711–716.
Discover more from Microbiology Class
Subscribe to get the latest posts sent to your email.