Introduction
Quality control (QC) is a systematic monitoring and verification framework used to detect, measure, and correct analytical and production errors by establishing predefined performance limits. It ensures that products and services consistently meet established quality specifications, regulatory standards, and customer expectations. In regulated industries such as pharmaceutical manufacturing, medical device production, and biotechnology, QC functions as a critical safeguard for product safety, efficacy, and consistency.
QC is implemented through structured testing procedures, performance verification, and continuous monitoring across all stages of production. It is particularly important in pharmaceutical and biological product manufacturing where contamination risks, microbial proliferation, and process variability can significantly impact product quality and patient safety.
Although quality control is closely related to quality assurance (QA), the two are distinct but complementary systems within a broader quality management framework.
Quality Control Versus Quality Assurance
Quality control focuses on operational activities that detect and correct defects in products or analytical results. It emphasizes product testing, inspection, measurement, and validation against established specifications.
Quality assurance, on the other hand, is a broader management system that ensures processes are designed and implemented to prevent errors before they occur. QA is preventive and system-oriented, whereas QC is detection-oriented and test-based.
In pharmaceutical manufacturing:
- QA ensures that standard operating procedures (SOPs), validation protocols, training programs, and documentation systems are properly designed and followed.
- QC verifies through laboratory and in-process testing that raw materials, intermediates, and finished products conform to required standards.
Both functions operate under Good Manufacturing Practice (GMP) requirements and are essential components of regulatory compliance.
Quality Control as a Core Component of Good Manufacturing Practice
Quality control is a fundamental element of Good Manufacturing Practice (GMP). Regulatory authorities require pharmaceutical manufacturers to implement robust QC systems to prevent contamination, ensure batch consistency, and maintain product integrity.
QC activities typically include:
- Raw material testing
- In-process control testing
- Environmental monitoring
- Microbiological testing
- Analytical verification of active pharmaceutical ingredients (APIs)
- Stability testing
- Final product release testing
A well-designed QC system ensures that no product is released to the market unless it has passed all required specifications. Batch release decisions must be supported by documented evidence generated through validated testing methods.
Raw Material Testing
Raw material testing is the first critical checkpoint in the quality control process. It ensures that all incoming materials—including active pharmaceutical ingredients (APIs), excipients, solvents, water, and packaging components—meet predefined identity, purity, potency, and safety specifications before use in production. Testing typically includes physical characterization, chemical assay, impurity profiling, and microbiological assessment where applicable. Verification of supplier certificates of analysis (CoA) is combined with independent laboratory testing to reduce reliance on external documentation. Proper raw material control prevents contamination, adulteration, and variability that could compromise product quality and patient safety.
In-Process Control Testing
In-process control testing involves monitoring critical parameters during manufacturing to ensure the process remains within validated operating limits. These tests assess variables such as pH, temperature, mixing time, particle size, weight variation, concentration, and uniformity at defined stages of production. Early detection of deviations allows immediate corrective actions before the batch progresses further. In-process controls reduce waste, prevent batch failure, and enhance process consistency. They also provide documented evidence that manufacturing conditions comply with established standard operating procedures (SOPs) and regulatory requirements under Good Manufacturing Practice (GMP) guidelines.
Environmental Monitoring
Environmental monitoring evaluates microbial and particulate contamination levels within manufacturing areas, particularly in sterile and controlled environments. It includes air sampling, surface swabbing, personnel monitoring, and water system testing to detect potential contamination sources. Results are compared with established alert and action limits to identify trends or deviations. Continuous monitoring ensures that cleanrooms, production lines, and aseptic processing areas maintain required classification standards. Effective environmental control reduces the risk of product contamination during exposure to the manufacturing environment. Documentation of monitoring results supports regulatory inspections and ongoing facility qualification.
Microbiological Testing
Microbiological testing ensures that pharmaceutical products, raw materials, and environments comply with microbial limit specifications. Testing typically includes enumeration of total viable microorganisms, detection of specified pathogens, sterility testing, and endotoxin or pyrogen assessment where applicable. These tests confirm that microbial contamination levels remain below acceptable safety thresholds. For non-sterile products, microbial limit testing verifies compliance with pharmacopeial standards. For sterile products, sterility assurance is critical. Accurate microbiological testing requires validated methods, qualified personnel, controlled laboratory conditions, and strict contamination prevention measures to maintain data reliability.
Analytical Verification of Active Pharmaceutical Ingredients (APIs)
Analytical verification of APIs confirms identity, purity, potency, and chemical integrity before incorporation into formulations. Testing includes assay determination, impurity profiling, residual solvent analysis, polymorphic characterization, and stability evaluation. Advanced analytical techniques such as high-performance liquid chromatography (HPLC), gas chromatography (GC), spectroscopy, and mass spectrometry are commonly employed. Verification ensures that APIs conform to pharmacopeial monographs or validated in-house specifications. Accurate API analysis prevents substandard or degraded materials from entering production. This step is essential for maintaining therapeutic efficacy, batch consistency, and regulatory compliance.
Stability Testing
Stability testing evaluates how pharmaceutical products maintain quality, potency, and safety over time under defined environmental conditions. Products are stored under accelerated and long-term conditions to assess degradation patterns, impurity formation, physical changes, and microbial growth potential. Stability data determine product shelf life, storage conditions, and packaging requirements. Testing typically follows international guidelines such as ICH recommendations. Continuous stability monitoring supports post-market surveillance and ensures that product performance remains consistent throughout its intended lifespan. Reliable stability studies are critical for regulatory approval and lifecycle management.
Final Product Release Testing
Final product release testing confirms that a finished batch meets all predefined quality specifications before it is authorized for market distribution. Tests include identity confirmation, potency assay, impurity analysis, microbial limits, sterility assessment (if applicable), and physical quality evaluation. Results are reviewed by qualified personnel to ensure compliance with approved product specifications and regulatory requirements. Batch release decisions are documented and traceable. No product may be distributed without passing final quality assessment. This control step provides the ultimate verification that manufacturing and testing processes have successfully produced a safe and effective pharmaceutical product.
Key Principles of a Sustainable Quality Control System
A sustainable QC system must be:
Practical
Testing procedures should align with the manufacturing scale, available laboratory infrastructure, and technical capacity. Overly complex testing systems that cannot be consistently implemented reduce reliability.
Achievable
QC procedures must be realistically executable within the operational context of the facility. Required equipment, trained personnel, validated methods, and reference standards must be available.
Affordable
Cost efficiency is essential. Laboratories should prioritize risk-based testing strategies that focus resources on critical control points rather than excessive redundant testing.
Balancing practicality, feasibility, and affordability ensures long-term sustainability of the QC framework.
Quality Control Across the Manufacturing Lifecycle
Quality control operates throughout procurement, processing, and product release rather than only at the finished stage. Continuous monitoring ensures deviations are detected early, risks are mitigated promptly, and product quality remains consistent across all production phases. Quality control activities occur at multiple stages of production rather than solely at the final product stage as seen below:
1. Raw Material Testing
Raw materials including active pharmaceutical ingredients, excipients, water systems, and packaging components must undergo verification before production use. This prevents contamination or substandard inputs from entering manufacturing processes and compromising final product quality.
Identity Verification
Identity testing confirms that each raw material matches its labeled specification using analytical methods such as spectroscopy, chromatography, or chemical reactions. This prevents substitution errors, mix-ups, or falsified materials from entering production.
Purity Assessment
Purity assessment quantifies the proportion of the desired compound relative to impurities or degradation products. It ensures materials meet defined chemical standards and reduces risks associated with toxic contaminants or inconsistent performance.
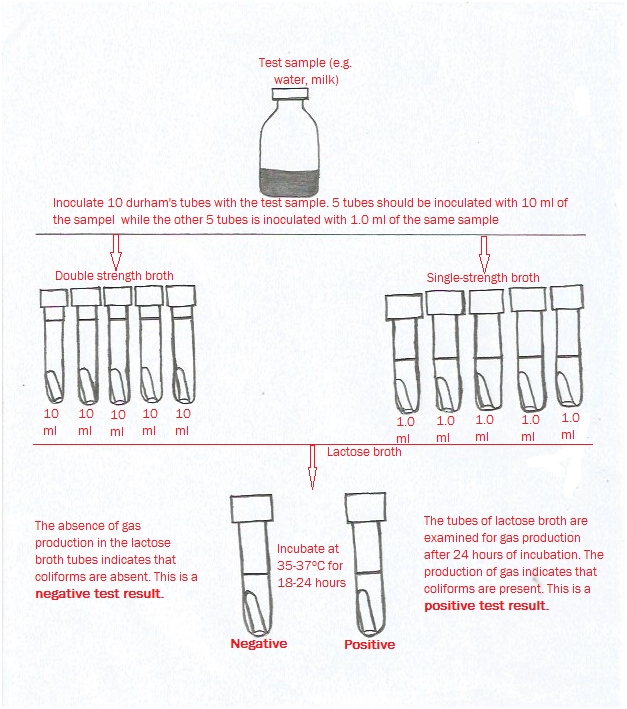
Microbial Limit Testing
Microbial testing determines the presence and level of viable microorganisms in raw materials. It is essential for materials prone to contamination and ensures bioburden levels remain within acceptable regulatory limits before use.
Endotoxin Screening
Endotoxin testing detects bacterial lipopolysaccharides that may cause fever or inflammatory reactions. It is critical for water systems and materials used in injectable or sterile product manufacturing to ensure patient safety.
Chemical Impurity Testing
Chemical impurity analysis identifies and quantifies residual solvents, heavy metals, degradation compounds, or process-related contaminants. Controlling impurities protects product efficacy, stability, and compliance with pharmacopeial standards.
Contaminated or non-conforming raw materials can compromise entire production batches, trigger costly recalls, and introduce safety hazards. Early detection through rigorous testing reduces downstream failures and strengthens overall manufacturing reliability.
2. In-Process Quality Control
In-process testing ensures that production parameters remain within defined control limits during manufacturing.
Examples of in-process control include:
Monitoring mixing time and temperature
Continuous supervision of mixing duration and thermal conditions ensures uniform blending, prevents degradation of heat-sensitive ingredients, and maintains formulation consistency within validated process parameters during production.
Measuring pH at defined intervals
Periodic pH measurement confirms chemical stability, optimal reaction conditions, and compatibility of ingredients. It detects deviations early, preventing product instability, reduced efficacy, or microbial growth during manufacturing.
Checking concentration during formulation
Verifying active ingredient concentration ensures accurate dosing, uniform distribution, and compliance with product specifications. Early detection of deviation prevents batch failure and guarantees therapeutic consistency.
Environmental monitoring of cleanrooms
Routine assessment of air, surfaces, and personnel in controlled areas detects microbial or particulate contamination. It ensures compliance with cleanliness standards and minimizes contamination risk in sterile production environments.
Sampling intermediate products for microbial contamination
Testing intermediates identifies contamination before final processing. Early microbial detection enables corrective action, reduces waste, and protects final product quality and patient safety.
In-process controls enable early intervention before deviations affect the finished product.
Continuous monitoring allows immediate correction of process errors, preventing propagation of defects. It enhances process control, reduces batch rejection risk, and ensures consistent product quality before release.
3. Environmental Monitoring
Pharmaceutical manufacturing environments especially sterile production areas must be continuously monitored for microbial contamination.
Environmental monitoring typically includes:
Air Sampling
Air sampling measures airborne microbial contamination within controlled manufacturing areas using active or passive sampling methods to detect microorganisms suspended in air and assess environmental cleanliness levels.
Surface Swabbing
Surface swabbing involves collecting samples from equipment, walls, floors, and work surfaces to detect microbial contamination. It verifies cleaning effectiveness and ensures surfaces remain within defined microbial limits.
Personnel Monitoring
Personnel monitoring assesses microbial contamination originating from operators through gown sampling, glove prints, and finger dabs. It evaluates hygiene compliance and minimizes contamination risk during aseptic operations.
Monitoring of Water Systems
Water system monitoring tests purified and water-for-injection systems for microbial load and endotoxins. It ensures water quality meets pharmaceutical standards and prevents contamination of formulations.
Evaluation Against Alert and Action Limits
Environmental results are compared to predefined microbial alert and action limits. Exceedances trigger structured investigations, root cause analysis, and corrective measures to restore controlled manufacturing conditions.
Results of environmental monitoring are compared with established microbial alert and action limits. Any deviation triggers investigation and corrective action.
4. Microbial Quality Control in Pharmaceutical Manufacturing
Microbial contamination is one of the most critical risks in pharmaceutical production. Microorganisms may enter products through contaminated raw materials, water systems, equipment, personnel, or air. Microbial contamination compromises product safety and efficacy. Contaminants can originate from poorly controlled environments, unsterilized equipment, raw materials, or human handling, making stringent monitoring and preventive controls essential throughout production.
Microbial QC focuses on detecting and controlling:
- Bacteria
- Fungi
- Yeasts
- Endotoxins
- Pyrogens
Bacteria
Bacteria are common environmental and process contaminants that can proliferate in nutrient-rich pharmaceutical materials. Microbial testing identifies viable bacterial load and ensures levels remain within acceptable regulatory limits to prevent infection risks.
Fungi
Fungi include molds and filamentous organisms that grow in moist environments. They can contaminate raw materials and facilities, causing product degradation and potential allergenic or toxic effects if not detected and controlled effectively.
Yeasts
Yeasts are single-celled fungi capable of rapid growth in liquid formulations and aqueous systems. Their detection in microbial testing ensures contamination is identified early to prevent spoilage and maintain product sterility or quality.
Endotoxins
Endotoxins are heat-stable components of the outer membrane of Gram-negative bacteria. They can persist even after bacterial death and cause severe inflammatory reactions, making sensitive detection essential for injectable and parenteral products.
Pyrogens
Pyrogens are fever-inducing substances derived mainly from microbial components. Quality control testing ensures that pharmaceutical products, particularly injectables, are free from pyrogenic contamination to protect patient safety and regulatory compliance.
Testing for Pyrogens and Endotoxins
Several pharmaceutical products including injectable drugs, vaccines, infusion fluids, and injection water are particularly vulnerable to contamination by pyrogens.
What Are Pyrogens?
Pyrogens are fever-inducing substances, often derived from bacterial cell wall components such as lipopolysaccharides (LPS) from Gram-negative bacteria. Even when bacteria are not viable, their endotoxins may remain and cause severe inflammatory reactions if introduced into the human body. Because of this risk, pyrogen testing is mandatory for parenteral products.
Common Pyrogen Testing Methods
- Rabbit Pyrogen Test
- Traditional biological method
- Measures temperature rise in rabbits after product injection
- Less commonly used due to ethical and variability concerns
- Limulus Amebocyte Lysate (LAL) Test
- Detects bacterial endotoxinsBased on clotting reaction from horseshoe crab blood
- Highly sensitive and widely used
- Recombinant Factor C (rFC) Assay
- Animal-free alternative to LAL
- Increasingly adopted in modern laboratories
QC laboratories must validate pyrogen testing methods and ensure proper calibration of equipment used for detection.
Control of Laboratory Materials and Equipment
Reliable QC results depend on strict control of laboratory resources.
Culture Media Control
All microbiological culture media must be:
- Sterility tested
- Growth promotion tested
- Properly stored
- Within expiry date
- Prepared under controlled conditions
Media performance must be verified using reference microbial strains to confirm growth support capability.
Reagent Control
Reagents used in chemical and microbiological analysis must:
- Meet specification requirements
- Be traceable to certified suppliers
- Be labeled with preparation and expiry dates
- Be stored under defined conditions
Improper reagent management increases analytical variability and error risk.
Equipment Calibration and Maintenance
Calibration ensures measurement accuracy and data integrity.
Calibration aligns instrument readings with recognized standards to guarantee reliable measurements. It reduces systematic error, supports traceability to reference standards, and strengthens data credibility. Accurate calibration is fundamental for regulatory compliance, scientific reproducibility, and consistent quality control outcomes across pharmaceutical and microbiological laboratories.
Routine calibration
Routine calibration involves comparing instrument measurements against certified reference standards at defined intervals to ensure accuracy and traceability. It detects measurement deviation caused by drift, wear, or environmental influence. Calibration schedules should be risk-based and documented. Any deviation beyond acceptable limits requires adjustment, correction, or repair to restore measurement reliability.
Preventive maintenance
Preventive maintenance consists of planned servicing activities designed to reduce equipment failure and unexpected downtime. It includes cleaning, inspection, lubrication, component replacement, and functional checks performed according to manufacturer recommendations. Regular maintenance extends equipment lifespan, enhances operational stability, and minimizes variability in analytical or sterilization processes.
Performance qualification (PQ)
Performance qualification verifies that equipment consistently operates according to predefined specifications under actual working conditions. It demonstrates that the instrument performs effectively within its intended use. PQ typically follows installation qualification (IQ) and operational qualification (OQ), and includes documented testing to confirm reproducibility, accuracy, and reliability of results.
Documentation of service records
Service documentation provides traceability and regulatory compliance evidence for all calibration, maintenance, repairs, and qualification activities. Records include dates, findings, corrective actions, calibration certificates, and technician signatures. Proper documentation supports audits, inspections, and investigations while ensuring transparency and accountability in equipment management.
Examples of Critical Laboratory Equipment that Requires Regular Calibration
Autoclaves
Autoclaves sterilize media, instruments, and laboratory waste using high-pressure saturated steam. Calibration ensures temperature, pressure, and time parameters achieve validated sterilization cycles. Routine monitoring with biological and chemical indicators confirms performance. Proper maintenance prevents leakage, temperature deviation, and incomplete sterilization that could compromise microbiological testing accuracy.
Incubators
Incubators maintain controlled temperature and environmental conditions for microbial growth and culture testing. Calibration verifies temperature uniformity and stability across chambers. Regular cleaning and validation ensure consistent growth conditions. Temperature fluctuations or contamination inside incubators can alter microbial test outcomes and affect quality control reliability.
Spectrophotometers
Spectrophotometers measure absorbance or transmittance for quantitative chemical and microbiological analyses. Calibration ensures wavelength accuracy, photometric precision, and baseline stability. Routine verification with certified standards maintains measurement consistency. Instrument drift or optical contamination can distort analytical data, making calibration critical for accurate laboratory results.
pH meters
pH meters measure acidity or alkalinity in solutions used for formulation and microbial media preparation. Calibration with standard buffer solutions ensures electrode accuracy and response stability. Regular electrode cleaning and replacement improve performance. Incorrect pH readings may affect product formulation, microbial growth conditions, and assay reliability.
Microbial counting systems
Microbial counting systems quantify viable microorganisms in samples using manual or automated methods. Calibration ensures accurate colony detection, image analysis, or fluorescence interpretation. Validation confirms reproducibility and sensitivity. Proper maintenance prevents counting errors that could misclassify contamination levels and compromise product safety assessments.
Laminar airflow cabinets
Laminar airflow cabinets provide controlled, particle-free airflow environments for aseptic operations and media preparation. Certification verifies airflow velocity, HEPA filter integrity, and particulate control. Routine testing ensures containment efficiency and sterility protection. Filter damage or airflow disruption increases contamination risk and undermines microbiological quality control processes.
Documentation and Record Keeping in Quality Control
Documentation is a cornerstone of GMP-compliant QC systems. It is a cornerstone of GMP-compliant QC systems.Documentation establishes evidence that quality control activities were performed according to approved procedures and specifications. It ensures accountability, consistency, and regulatory compliance. Proper documentation captures all testing activities, decisions, and outcomes, forming an auditable trail that demonstrates product quality and process control throughout the manufacturing lifecycle.
Essential records include:
- Test protocols
- Raw data sheets
- Instrument calibration logs
- Batch testing reports
- Deviation reports
- Corrective and preventive action (CAPA) records
Accurate documentation provides traceability and supports regulatory inspections. Data integrity must be preserved through controlled record management systems.
Test protocols
Test protocols define the methodology, acceptance criteria, sampling plan, and analytical procedures used in quality control testing. They provide standardized instructions for laboratory personnel to follow, ensuring reproducibility and consistency. Approved protocols reduce variability in testing outcomes and ensure that results are scientifically valid and compliant with regulatory expectations.
Raw data sheets
Raw data sheets capture original observations and measurements generated during laboratory testing. These include instrument readings, calculations, chromatograms, microbial counts, and observations recorded at the time of analysis. Raw data must be accurate, traceable, and unaltered to preserve data integrity and allow verification of reported results during audits or inspections.
Instrument calibration logs
Calibration logs document the verification of instrument accuracy against certified standards at defined intervals. They record calibration dates, results, deviations, adjustments, and maintenance actions. These logs demonstrate that equipment used for testing operates within acceptable limits and produces reliable measurements, supporting confidence in analytical data.
Batch testing reports
Batch testing reports summarize the results of quality control tests performed on a specific production batch. They compare analytical findings against predefined acceptance criteria and confirm compliance. These reports form the basis for batch release decisions and provide documented evidence that the manufactured product meets required quality standards.
Deviation reports
Deviation reports document any departure from approved procedures, specifications, or expected results during manufacturing or testing. They describe the nature of the deviation, root cause analysis, investigation findings, and impact assessment. Proper documentation ensures deviations are evaluated systematically and corrective measures are implemented to prevent recurrence.
Corrective and preventive action (CAPA) records
CAPA records capture actions taken to correct identified problems and prevent their recurrence. Corrective actions address immediate issues, while preventive actions mitigate potential future risks. Documentation includes investigation results, implemented solutions, effectiveness checks, and follow-up verification, ensuring continuous improvement within the quality system.
Accurate documentation and data integrity
Accurate documentation ensures traceability from raw materials to finished products and supports regulatory inspections and audits. Data integrity requires that records are complete, consistent, attributable, and secure. Controlled record management systems whether paper-based or electronic protect data from unauthorized alteration and maintain transparency in quality control operations.
Batch Release and Product Disposition
Finished pharmaceutical products must not be released to the market until:
- All required QC tests are completed
- Results meet predefined specifications
- Deviations are investigated and resolved
- Qualified personnel authorize release
Batch release is a formal decision supported by documented QC evidence. Unauthorized product release violates GMP regulations and compromises patient safety and that of the general public.
Risk-Based Approach to Quality Control
Modern QC systems increasingly adopt a risk-based strategy. A risk-based quality control approach aligns testing intensity and monitoring efforts with the potential impact of a defect on product safety, efficacy, and regulatory compliance. Instead of applying uniform testing to all materials and processes, resources are allocated according to scientific assessment of risk. This approach is consistent with current GMP principles and international regulatory expectations. It improves laboratory efficiency, reduces unnecessary testing, and strengthens oversight of critical production steps. By focusing on areas that pose the greatest threat to product quality, manufacturers achieve better control with optimized operational cost and compliance performance. Risk-based QC improves efficiency while maintaining safety standards. As aforesaid, modern QC systems increasingly adopt a risk-based strategy. This approach involves:
Identifying critical quality attributes (CQAs)
Critical quality attributes are physical, chemical, biological, or microbiological characteristics that must remain within defined limits to ensure product safety and effectiveness. Identification of CQAs begins during product development and process validation. Examples include potency, sterility, purity, particle size, and endotoxin levels for injectable products. Risk assessment tools such as hazard analysis and critical control point (HACCP), failure mode and effects analysis (FMEA), and process capability studies are often used to determine which attributes are most critical. Clear definition of CQAs ensures that quality control testing focuses on measurable parameters that directly affect patient safety and therapeutic performance.
Evaluating risk probability and severity
Risk evaluation involves assessing both the likelihood of a quality failure occurring and the potential consequences if it occurs. Probability considers factors such as process complexity, historical deviations, supplier reliability, and environmental conditions. Severity evaluates the impact on patient health, product efficacy, and regulatory compliance. A structured scoring system is often used to classify risks as low, medium, or high. This quantitative or semi-quantitative analysis supports objective decision-making. By combining probability and severity, manufacturers can prioritize control measures for processes that present the highest overall risk to product integrity and public health.
Prioritizing testing based on product risk classification
Once risks are assessed, products and processes are categorized according to their risk level. High-risk products, such as sterile injectables or biologics, require more rigorous and frequent testing compared to low-risk oral solid dosage forms. Testing frequency, sampling size, and analytical depth are adjusted based on this classification. Critical materials from high-risk suppliers may undergo enhanced verification, while well-established suppliers with strong quality histories may require reduced testing under controlled conditions. This prioritization ensures that laboratory capacity is used efficiently while maintaining adequate oversight of parameters that directly influence product quality and compliance.
Implementing enhanced monitoring for high-risk products
High-risk products require intensified monitoring throughout the manufacturing lifecycle. Enhanced monitoring includes increased environmental surveillance, tighter in-process control limits, additional microbial testing, and more frequent equipment validation. Real-time monitoring technologies and automated data systems are often deployed to detect deviations early. Continuous trend analysis helps identify emerging risks before they escalate into product failures. Corrective and preventive actions (CAPA) are implemented rapidly when deviations occur. This proactive surveillance reduces the likelihood of contamination or process drift. Ultimately, enhanced monitoring strengthens product assurance, ensures regulatory readiness, and reinforces confidence in products with significant patient safety implications.
Quality control is a vital monitoring system that safeguards pharmaceutical product quality through systematic testing, validation, and verification. It detects and corrects analytical errors, ensures compliance with defined standards, and prevents contaminated or substandard products from reaching consumers.
In pharmaceutical manufacturing, QC spans raw material testing, in-process monitoring, environmental surveillance, microbial testing, pyrogen detection, and final product verification. Effective control of laboratory materials including culture media, reagents, and equipment ensures reliability and accuracy of test results.
A sustainable QC framework must be practical, achievable, and affordable while aligned with GMP requirements and regulatory expectations. When properly implemented, quality control protects public health, enhances manufacturing consistency, and strengthens trust in pharmaceutical products.
References
Arora D.R (2004). Quality assurance in microbiology. Indian J Med Microbiol, 22:81-86.
Ashutosh Kar (2008). Pharmaceutical Microbiology, 1st edition. New Age International Publishers: New Delhi, India.
Axelsen P.H (2002). Essentials of antimicrobial pharmacology. Humana Press, Totowa, New Jersey, USA. Al-Jasser A.M (2006). Extended – Spectrum Beta – Lactamases (ESBLs): A Global Problem. Kuwait Medical Journal, 38(3):171-185.
Bisht R., Katiyar A., Singh R and Mittal P (2009). Antibiotic Resistance – A Global Issue of Concern. Asian Journal of Pharmaceutical and Clinical Research, 2 (2):34-39.
Block S.S (2001). Disinfection, sterilization and preservation. 5th edition. Lippincott Williams & Wilkins, Philadelphia and London.
Cars O and Nordberg P (2005). Antibiotic resistance: The faceless threat. International Journal of Risk & Safety in Medicine, 17 (3/4): 103-110.
Carson C.F., Hammer K.A and Riley T.V (2006). Malaleuca alternifolia (Tea Tree) oil: A Review of Antimicrobial and other Medicinal Properties. Clinical Microbiology Review, 19(1):50-62.
Cowan M.M (1999). Plant products as antimicrobial agents. Clinical Microbiology Reviews., 564-582.
Denyer S.P., Hodges N.A and Gorman S.P (2004). Pharmaceutical Microbiology. 7th ed. Blackwell Publishing Company, USA.
Finch R.G, Greenwood D, Norrby R and Whitley R (2002). Antibiotic and chemotherapy, 8th edition. Churchill Livingstone, London and Edinburg.
Joslyn, L. J. (2000). Sterilization by Heat. In S. S. Block (Ed.), Disinfection, Sterilization, and Preservation (5th ed., pp. 695-728). Philadelphia, USA: Lippincott Williams and Wilkins.
Lai P.K and Roy J (2004). Antimicrobial and chemopreventive properties of herbs and spices. Curr. Med. Chem, 11 (11): 1451–1460.
Livermore D.M (2004). The need for new antibiotics. Clinical Microbiology & Infection, 4(10): 1-9.
Mascaretti O.A (2003). Bacteria versus antibacterial agents: An integrated approach. Washington: ASM Press.
Nally J.D (Ed.) (2007). Good manufacturing practices for pharmaceuticals. Sixth edition. Informa Healthcare USA, Inc, New York.
Discover more from Microbiology Class
Subscribe to get the latest posts sent to your email.