
PCR is the acronym for “polymerase chain reaction”. Polymerase chain reaction is the molecular biology technique that is used to amplify or copy specific gene sequences or nucleotides of a DNA molecule or gene. It was invented by Kary Mullis and his colleagues in the early 1980s; and PCR technique is the most widely used nucleic acid amplification technique that is used to make several copies of a given gene sequence.
PCR technique is also the gold standard for a variety of amplification processes in the molecular biology laboratory, in diagnostic institutions such as hospitals and in research and academic institutions. PCR technique is generally an in vitro procedure that allows scientists to amplify a target DNA sequence (often known as the gene of interest) as well as make several copies of it under controlled heating and cooling processes made possible by the thermocycler (Figure 1).
The thermocycler is like the backbone of the PCR technique because without this piece of equipment; gene sequences or DNA might not be copied and amplified for further genetic manipulations or study. The use of PCR techniques for several medical and diagnostic practices blossomed due to the discovery of specific enzymes (particularly Taq polymerase) that allows the gene of interest to be amplified in vitro by certain enzymatic and replicative reactions.
Taq polymerase was initially isolated from the hot spring bacterium, Thermus aquaticus; and this enzyme have been playing vital role in many PCR technologies. The Taq polymerase (Taq Pol) enzyme replaced DNA polymerase enzyme isolated from Escherichia coli, and previously used for PCR experimentation; and Taq Pol or Taq is able to withstand the high temperature conditions required during PCR techniques than the DNA polymerase enzymes. PCR technique generally involves a series of heating or melting processes that occur at high temperature, and it is critical that enzymes such as the Taq polymerase which can withstand such a high temperature are available and used for the successful amplification of a particular gene sequence.
Man’s understanding about the macromolecules of live (particularly DNA) have been greatly improved and advanced due to the possibility of analyzing the gene sequences of different organisms or living systems at even a shorter period of time. Sufficient amount of the gene of interest of a given organism can be isolated, copied and analyzed or manipulated to generate a given gene product because of the application of PCR technologies.
The PCR technique involves a series of thermal cycling of the reaction mixture in specialized tubes known as Eppendorf tubes (Figure 2); and this process consists of different controlled cycles of repeated heating and cooling of the reaction mixture in the tube for DNA melting and further enzymatic replication of the DNA of interest or a particular gene sequence. PCR technique has the advantage of speed and sensitivity for molecular studies.

Thermocycler machine is used to conduct PCR using defined primers, defined DNA of test organism or sample and defined temperatures for specific time intervals. PCR is an important technique in molecular biology; and it is used to achieve many purposes in the laboratory such as for the amplification of gene or DNA and gene cloning. Standard aseptic techniques applied in microbiological experimentations should also be applied to PCR experiments in order to forestall any risk of possible contamination during the process.

COMPONENTS OF A PCR REACTION MIXTURE
PCR techniques cannot be performed the same way other routine microbiological procedures are carried out. It is not always a routine technique in most hospitals except in cases where they are applied for advanced research works. However, the PCR technique is almost a routine in advanced molecular biology laboratories. There are some components that make the process to be successful. The PCR technique occurs in a reaction mixture contained in specialized tubes known as the PCR tube or Eppendorf tubes; and the components of the reaction are mixed at varying concentrations in these tubes. The main component of a PCR technique are:
- DNA template: The DNA template is the macromolecule that contains the DNA region or gene sequence to be copied and amplified. It is usually isolated from the organism and later purified by DNA isolation and purification techniques prior to their usage as template DNA.
The template DNA is obtained as a part from the whole chunk of the organism’s genomic DNA; and it is the part that contains the target DNA sequence or gene of interest to be amplified. RNA can also serve as a template DNA for a PCR technique, but DNA is more preferable in most PCR techniques because they are more stable than RNA; and the isolation of DNA is much simpler than isolating RNA.
- Primers: To begin the synthesis of a new DNA strand, a short fragment of DNA or RNA known as the primer, must be created and paired with the template DNA strand before the Taq polymerase enzyme can begin the actual process of synthesizing the new DNA strand.
Primers are short DNA fragments (usually about 18-20 bases or nucleotides long) that contain specific sequences which are complementary to the template DNA molecule to be copied and amplified. Two types of primers (i.e. the forward primers and the reverse primers) that are complementary to the 3′ end of each of the double stranded template DNA strands are used in any PCR reaction process.
Forward primers can also be called sense primers while the reverse primers are also known as the anti-sense primers. Primers generally bind to the opposite sides of the template DNA sequence or strands to be amplified during the PCR reaction. They are single-stranded (ss) DNA molecules or oligonucleotides, and they are synthesized by chemical processes.
- Taq polymerase enzyme: Taq polymerase (Taq Pol) enzyme is a thermostable enzyme that is used in PCR techniques to copy DNA molecules. DNA polymerase also performs the same function as the Taq Pol enzyme. They bind to ss-DNA molecules (particularly the primers) and synthesize new DNA strands that are complementary to the template DNA molecule. Taq polymerase enzyme is generally used in PCR reaction because of their ability to withstand the high temperatures (up to 100oC) that is prevalent in the PCR reaction process. They specifically add dNTPs to the 3′ OH terminus of a growing DNA chain.
- Nucleotides or dNTPs: dNTPs is the acronym for “deoxynucleosides triphosphate”; and they are nucleotides that contain triphosphate groups. Nucleotides or dNTPs are generally the main building blocks from which the DNA polymerase or Taq Pol enzyme synthesizes a new DNA strand. The dNTPs are derived from the bases that make up the DNA which are adenine (A), guanine (G), cytosine (C) and thymine (T).
Four dNTPs or nucleosides exist, and they include deoxyadenosine triphosphate (dATP), deoxycytidine triphosphate (dCTP), deoxyguanosine triphosphate (dGTP), and deoxythymidine triphosphate (dTTP), and they are individually added to the new DNA strand by the Taq Pol enzyme during the PCR reaction process.
- Buffer solution: A buffer is a substance that keeps a constant balance between an acid and a base or alkali in a solution. Buffers are used to maintain equilibrium between basic and acidic solutions. In PCR reactions, buffers are usually added to the reaction mixture in order to provide a suitable environment for optimum reaction and stability of the Taq Pol and other reaction mixtures. Buffers are generally used to maintain constant pH levels for PCR reaction processes; and the optimal pH level of most PCR reactions is usually in the range of 8.3 – 9.0.
- Divalent cations: Divalent cations such as magnesium ions (Mg2+) in the form of magnesium chloride (MgCl2) are also included in the PCR reaction mixture; and they act as enzyme cofactors during the PCR reaction process.
There are several types of PCR techniques used by molecular biologists to study the gene sequences of a given DNA molecule or gene of interest. Real-Time (RT) PCR, simple PCR and nested PCR are some of the known PCR techniques used in molecular biology, but the type of PCR technique used is usually informed by the type of investigation to be undertaken; and the expertise of the investigator is critical to the success of the process.
The polymerase chain reaction is widely used in molecular biology, microbiology, genetics, diagnostics clinical laboratories, forensic science, and environmental science amongst other biological, medical and biomedical sciences to study living systems at the molecular level. PCR technique is applied in the detection and diagnosis of infectious diseases; in paternity testing; for DNA sequencing or gene cloning; and for identification of genetic fingerprints to solve some cases of crimes.
PCR technique is a very sensitive experiment, and thus it should be carried out in clean environment (e.g. in biosafety laminar flow cabinet) to avoid contaminating the process with exogenous DNA’s from the laboratory environment. And all materials required for the process must be kept clean and away from any possible source of contamination.
STEPS INVOLVED IN PCR TECHNIQUE
PCR technique usually involves three stages of cycling which is denaturation, renaturation and elongation step. All these stages of DNA amplification are carried out at different temperatures that involve repeated cycles of incubation which occurs in the reaction tube in the thermocycler.
- Denaturation: Denaturation is the first stage in a PCR reaction, and it is the stage at which a double stranded (ds) DNA molecule is unwounded, or separated into their individual complementary single strands. At this stage, the PCR reaction mixture is heated to a high temperature (about 94-98oC) for 20-30 seconds. This high temperature is necessary for the strand separation of the template dsDNA molecule, without which there will not be any form of copying and amplification of the gene of interest or DNA by the Taq polymerase enzyme. Denaturation breaks down the hydrogen bonds involved in base pairing of dsDNA molecules.
- Renaturation (annealing): Renaturation is the second stage of the PCR reaction. This stage is also known as annealing, and it is the stage in the PCR reaction in which the reaction process is rapidly cooled to a temperature of around 50-65oC for 20-40 seconds. Renaturation is generally the stage at which the primers (forward and reverse) anneal or hybridize to the complementary ssDNA template molecule. Complementarity is the ability of single-stranded DNA molecule to base-pair or anneal again after being denatured. The synthesis and/or replication of the new DNA strand actually begin at this stage of annealing. Once the primers are annealed to the template DNA, the Taq Pol enzyme initiates the process of elongation of the DNA molecule. The cooling temperature obtainable in this stage is suitable for the effective binding of the primers to the template DNA.
- Elongation: Elongation or extension is the third stage in the PCR reaction process. In this stage, the PCR reaction mixture is heated to a temperature of 72oC for primer extension. The Taq Pol enzyme is most active in this stage because the ongoing DNA synthesis is extended and elongated into new DNA strands. The dNTPs required for building the new DNA strands are continuously added in the 5′-3′ direction at this stage of elongation. At this stage, the amplification of the gene of interest occurs at an exponential state, and several copies of the desired DNA (in thousands or millions) are produced or synthesized at the end of the whole cycling process.
While the temperatures of denaturation and elongation may remain constant for a PCR reaction, the temperature required for the annealing or renaturation stage usually varies with the type of PCR technique or analysis to undertake and this difference is usually due to the guanine-cytosine (G+C) content of the primers used for the assay. G+C content of primers varies. Additional repeated cycles of incubation of the PCR reaction in the thermocycler leads to the synthesis of many copies of the gene of interest or target DNA (Figure 3).
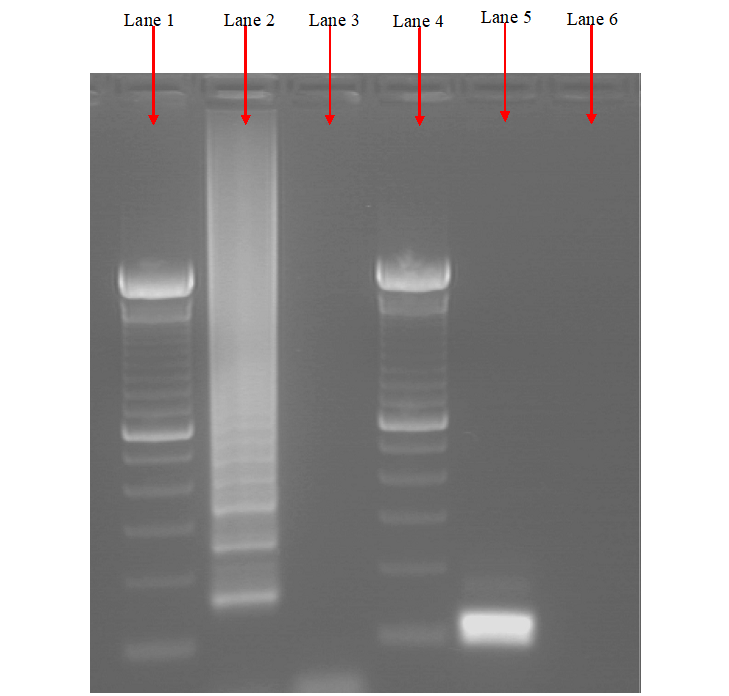
The products of a PCR reaction are run in a gel electrophoresis apparatus which generates the separated DNA fragments according to their different sizes and charges when passed through an electric current. After gel electrophoresis, the DNA fragments on the gel slab is photographed under a UV light (after been stained with EtBr), and it is compared to a standard DNA molecular marker to determine the actual sizes of the DNA fragments usually expressed in kilobase (Figure 4).
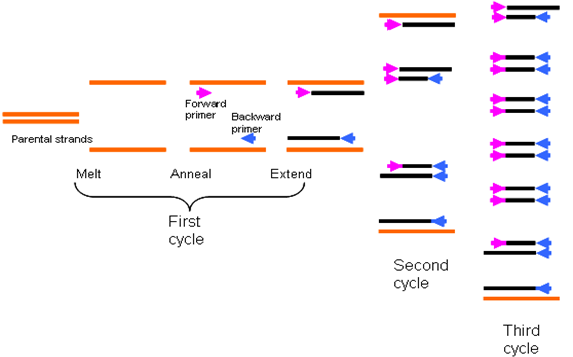
The number of cycles required for optimum amplification of a particular gene varies; and this is usually dependent on the amount of the starting material. In some PCR techniques, cycles can range from 25-40 cycles, but the longer the cycle the possibility of the accumulation of nonspecific nucleic acid products. Thus, the number of cycles to include in your PCR technique should be tailored down to the smallest number of cycles required to generate multiples copies of the gene sequence of interest as desired by the researcher. With PCR technique, many copies of a particular gene sequence of nucleic acid (ranging in millions) can be generated from only a few copies of the original DNA through amplification of the gene at different cycles.
References
Alberts B, Bray D, Lewis J, Raff M, Roberts K and Watson J.D (2002). The molecular Biology of the Cell. Fourth edition. New York, Garland, USA.
Chen I and Dubnau D (2004). DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2 (3): 241–249.
Cooper G.M and Hausman R.E (2004). The cell: A Molecular Approach. Third edition. ASM Press.
Dale J (2003). Molecular genetics of bacteria. Jeremy W. Dale and Simon Park (4th eds.). John Wiley & Sons Ltd, West Sussex, UK. Pp. 312-313.
Das H.K (2010). Textbook of Biotechnology. Fourth edition. Wiley edition. Wiley India Pvt, Ltd, New Delhi, India.
Lewis R (2004). Human Genetics: Concepts and Applications. Sixth edition. McGraw Hill Publishers, USA.
Lodish H, Berk A, Matsudaira P, Kaiser C.A, Kreiger M, Scott M.P, Zipursky S.L and Darnell J (2004). Molecular Cell Biology. Fifth edition. Scientific American Books, Freeman, New York, USA.
Madigan M.T., Martinko J.M., Dunlap P.V and Clark D.P (2009). Brock Biology of Microorganisms, 12th edition. Pearson Benjamin Cummings Inc, USA.
McPherson M and Moller S (2002). PCR: The Basics. 2nd edition. Taylor and Francis Group. New York, USA.
Sambrook, J., Russell, D.W. (2001). Molecular Cloning: a Laboratory Manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York.
Synder L, Peters J.E, Henkin T.M and Champness W (2013). Molecular Genetics of Bacteria. Fourth edition. American Society of Microbiology Press, USA.
Tamarin Robert H (2002). Principles of Genetics. Seventh edition. Tata McGraw-Hill Publishing Co Ltd, Delhi.
Discover more from Microbiology Class
Subscribe to get the latest posts sent to your email.

