Confocal microscopy is defined as an optical imaging technique that enhances optical resolution and contrast by focusing light on a single point within a defined focal plane, thereby minimizing image distortion and allowing for the reconstruction of three-dimensional images from two-dimensional scans. It is an advanced optical imaging technique developed to enhance image resolution, contrast, and clarity by selectively eliminating out-of-focus light from specimens. Unlike conventional widefield microscopy, where fluorescence or reflected light from multiple focal planes contributes to image formation and often produces blurred images, confocal microscopy uses point illumination and a spatial pinhole aperture to ensure that only light originating from the focal plane is detected. This principle of optical sectioning allows researchers to obtain highly detailed images of thick biological and material specimens with exceptional spatial resolution.
The technique has become an indispensable tool across numerous scientific disciplines, including cell biology, microbiology, neuroscience, materials science, environmental science, and biomedical research. Its ability to generate thin optical slices of specimens enables the reconstruction of three-dimensional structures, providing valuable insights into cellular architecture, tissue organization, microbial biofilms, and intracellular processes. Confocal microscopy is especially valuable in fluorescence imaging, where fluorescent dyes, proteins, or probes are used to label specific molecules or structures within a sample. By combining fluorescence labeling with precise optical filtering, confocal systems produce images with significantly reduced background noise and improved signal specificity.
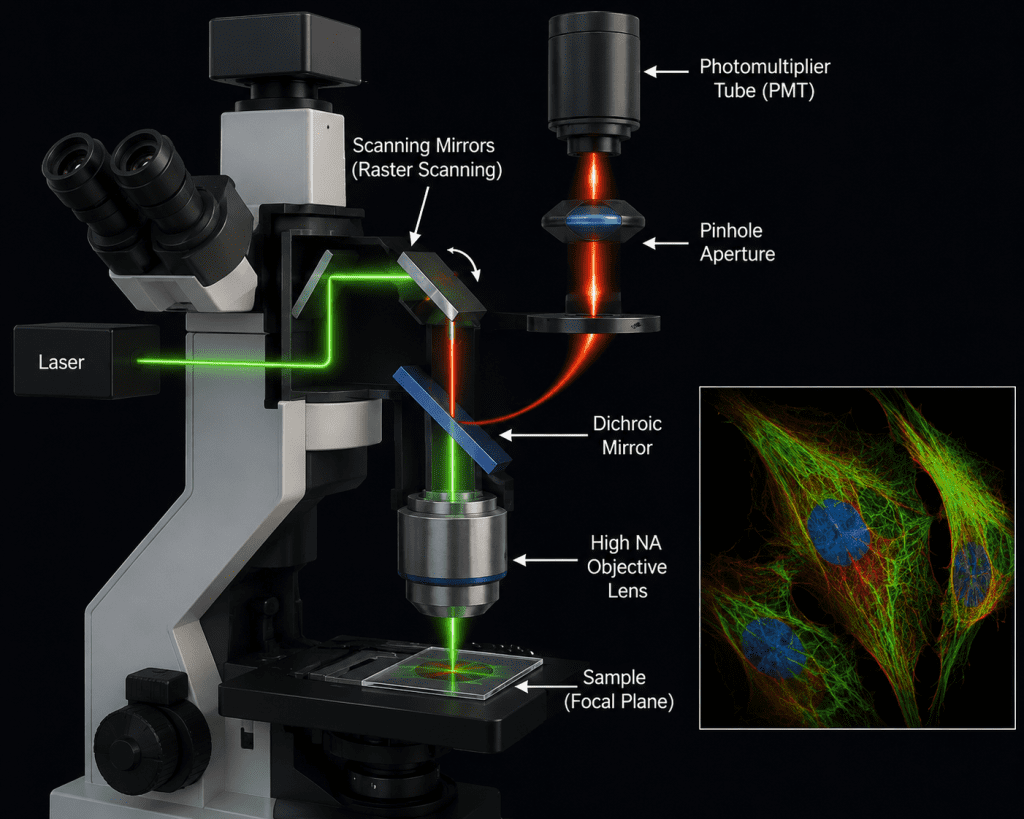
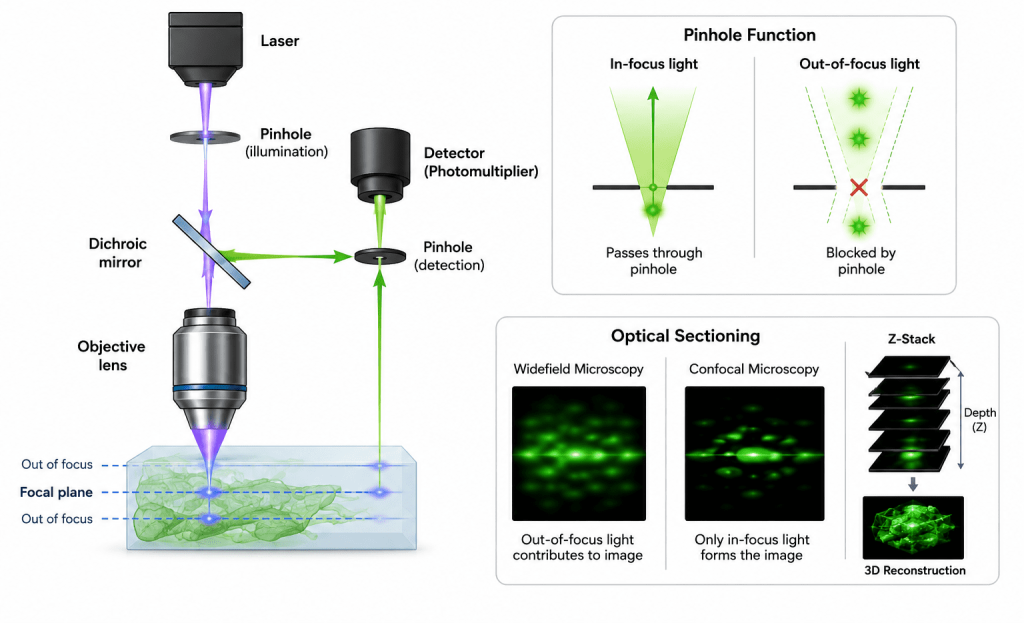
A confocal microscope typically consists of lasers for excitation, scanning mirrors for raster scanning, high numerical aperture objective lenses, dichroic mirrors, pinhole apertures, sensitive detectors such as photomultiplier tubes, and advanced computer software for image acquisition and analysis (Figure 1). Modern confocal systems can perform multichannel imaging, live-cell imaging, time-lapse analysis, and spectral imaging, thereby enabling researchers to study dynamic biological processes in real time. As aforesaid, a confocal microscope consists of several integrated optical and electronic components that work together to produce high-resolution images. The laser light source provides intense monochromatic light used to excite fluorophores within the specimen.
A dichroic beam splitter reflects the excitation light toward the sample while allowing emitted fluorescence to pass toward the detector. The objective lens, usually with a high numerical aperture, focuses the laser onto a precise point in the specimen and collects emitted light. A scanning system, typically made of galvanometric mirrors, moves the laser beam across the sample point by point to generate the image. The defining feature of the system is the pinhole aperture, positioned in front of the detector, which blocks out-of-focus light and enables optical sectioning. The emitted fluorescence that passes through the pinhole is detected by sensitive devices such as photomultiplier tubes (PMTs) or hybrid detectors, which convert light into electrical signals. Finally, a computer with specialized imaging software reconstructs these signals into detailed two-dimensional or three-dimensional images for visualization and analysis.
One of the most significant advantages of confocal microscopy is its capacity for optical sectioning and three-dimensional reconstruction. Sequential images collected at different focal depths, known as z-stacks, can be digitally reconstructed into volumetric models, allowing detailed visualization of complex biological and structural arrangements. This capability has revolutionized studies involving tissue morphology, microbial communities, and subcellular localization of proteins.
Despite its many advantages, confocal microscopy also has some limitations, including photobleaching, phototoxicity, relatively slow image acquisition in point-scanning systems, and limited imaging depth in highly scattering tissues. Nevertheless, continuous technological advancements such as spinning-disk confocal microscopy, multiphoton microscopy, adaptive optics, and AI-assisted image processing continue to improve imaging speed, sensitivity, and resolution.
Confocal microscopy represents a cornerstone imaging technology that bridges conventional light microscopy and modern high-resolution imaging. Its precision, versatility, and compatibility with fluorescence techniques make it an essential analytical tool in contemporary scientific and medical research, including emerging fields such as antimicrobial resistance studies, environmental microbiology, and live-cell functional imaging.
Principle of confocal microscopy
Confocal microscopy is an advanced optical imaging technique that enhances image resolution and contrast by selectively collecting light only from a specific focal plane within a specimen. The core principle underlying confocal microscopy is the combination of point illumination and a spatial pinhole aperture positioned in a plane conjugate to the focal plane of the specimen, which gives the technique its name “confocal.” Unlike conventional light microscopy, where light from multiple depths contributes to the final image, confocal microscopy eliminates most of the out-of-focus fluorescence or reflected light. As a result, it produces sharper, clearer, and higher-resolution images, especially when imaging thick biological samples such as tissues, biofilms, or multicellular structures.
The technique works by focusing a highly intense laser beam onto a very small diffraction-limited spot within the sample (Figure 2). Instead of illuminating the entire specimen simultaneously, the laser scans point-by-point across the sample in a raster pattern. Fluorescent light emitted from the illuminated point is then directed back through the optical system toward a detector. Before reaching the detector, the emitted light must pass through a small pinhole aperture. This pinhole blocks unfocused or scattered light originating from regions above or below the focal plane, thereby allowing only in-focus light to contribute to image formation.
The fundamental imaging principles of confocal microscopy are:
- Optical sectioning
One of the most important capabilities of confocal microscopy is optical sectioning. In traditional widefield microscopy, the whole specimen is illuminated at once, and fluorescence emitted from all layers of the sample is collected simultaneously. In thick specimens, this causes substantial background blur because out-of-focus light overlaps with the focal image plane. Consequently, image contrast and spatial resolution are significantly reduced.
Confocal microscopy overcomes this limitation through three key mechanisms:
- Focusing a laser beam onto a single diffraction-limited point
- Sequentially scanning this point across the specimen
- Using a pinhole aperture to reject out-of-focus light
Because only light from the focal plane is detected, the microscope can acquire very thin optical slices from different depths of the specimen. These optical sections are typically between 0.3 and 1.0 µm thick, depending on the wavelength, numerical aperture, and pinhole size. By collecting multiple optical sections along the z-axis, researchers can reconstruct detailed three-dimensional images of cells, tissues, microbial biofilms, or intracellular structures.
- Confocal aperture (Pinhole)
The pinhole aperture is the defining component of the confocal microscope and is essential for its improved imaging performance. It is positioned directly in front of the detector in a plane that is optically conjugate to the focal plane of the specimen. Its main function is to eliminate out-of-focus light before detection.
The size of the pinhole has a major influence on image quality:
- Smaller pinhole: improves axial resolution and optical sectioning but reduces signal intensity because less light reaches the detector.
- Larger pinhole: allows more light collection and increases brightness, but decreases axial resolution and permits more background blur.
Selecting the optimal pinhole diameter requires balancing image brightness against spatial resolution.
- Resolution
Confocal microscopy provides enhanced spatial resolution compared with conventional fluorescence microscopy. Typical values include:
- Lateral (x-y) resolution: approximately 200 nm
- Axial (z) resolution: approximately 500-700 nm
The resolving power of the system depends primarily on:
- The wavelength of the excitation light (λ)
- The numerical aperture (NA) of the objective lens
- The refractive index of the imaging medium
Shorter wavelengths and higher numerical apertures generally improve resolution. Consequently, high-NA oil or water immersion objectives are commonly used in confocal imaging to maximize image clarity and detail.
Image formation process in confocal microscopy
In confocal microscopy, the image is formed sequentially as follows:
- Laser illuminates a single point
- Fluorescence emitted from focal plane
- Out-of-focus light blocked by pinhole
- Detector records intensity
- Beam scans across sample
- Image reconstructed pixel by pixel
This process is repeated for different focal planes to generate a z-stack (Figure 3).
In confocal microscopy, image formation is fundamentally a sequential point-by-point optical sampling process, rather than the parallel illumination used in widefield microscopy. This scanning-based architecture is what enables optical sectioning and high-contrast imaging of thick specimens. The process begins with a highly focused laser beam that is directed through the objective lens to illuminate a single, diffraction-limited point within the specimen. Unlike conventional illumination, only a very small volume essentially a single voxel is excited at any given time. This localized excitation is critical for improving spatial resolution and reducing background fluorescence.
At the illuminated focal point, fluorophores within the specimen absorb energy from the laser and subsequently emit fluorescence photons. These emitted photons originate primarily from the focal plane, although some signal may also arise from above and below this plane due to scattering and out-of-focus excitation. A key feature of confocal imaging is the presence of a pinhole aperture positioned in a plane conjugate to the focal plane of the specimen. This pinhole acts as a spatial filter: it selectively allows photons originating from the focal point to reach the detector while effectively rejecting out-of-focus light. As a result, background blur is significantly reduced, and optical sectioning is achieved.

The transmitted fluorescence signal is then captured by a highly sensitive detector, such as a photomultiplier tube (PMT) or hybrid detector. The detector converts the incoming photons into an electrical signal proportional to fluorescence intensity at that specific spatial location. To construct a complete image, the laser beam is systematically scanned across the specimen in a raster pattern using galvanometric or resonant mirrors. At each coordinate position (x, y), the emitted intensity is measured and assigned to a corresponding pixel in the final image. This sequential acquisition means that a single optical slice is built pixel by pixel, rather than captured instantaneously.
Once a full two-dimensional plane is acquired, the focal position is incrementally shifted along the z-axis using precise mechanical or piezoelectric stage control. The entire scanning process is then repeated at successive depths within the sample. The collection of these optical sections forms a z-stack, which can be computationally reconstructed into a three-dimensional representation of the specimen. This enables visualization of internal structures, spatial relationships, and volumetric organization that are inaccessible in conventional microscopy. Confocal image formation integrates point illumination, spatial filtering, and synchronized scanning to produce high-resolution, depth-resolved digital images.
Z-stack phenomenon in confocal microscopy
A z-stack is a series of images taken at different focal depths (along the z-axis) of a specimen using a microscope, most commonly a confocal microscope. Instead of capturing just one flat image, the microscope acquires multiple “optical slices” through the sample:
- Each image corresponds to a specific depth (z-position)
- Together, they form a stack of 2D images (Figure 3)
So why is the z-stack used? It should be noted that biological samples are often thick and three-dimensional. Therefore, a single image cannot represent internal structure. A z-stack allows you to:
- Reconstruct a 3D representation of the specimen
- Analyze structures at different depths (e.g., cells inside tissues or biofilms)
- Measure spatial relationships in three dimensions
How does it work in confocal microscopy?
- The microscope focuses on the top plane → captures image
- The focus is moved slightly deeper (e.g., 0.2–1 µm step)
- Another image is captured
- This process repeats through the entire sample thickness
Output: The resulting dataset is a stack of images, which can be:
- Viewed slice-by-slice
- Rendered into a 3D model
- Processed for volume or structure analysis
Think of the z-stack like slicing a loaf of bread; each slice is one optical section (one image) while the full loaf is the complete z-stack (3D structure).
Types of confocal microscopy
Confocal microscopy is not a single unified instrument but a family of related imaging architectures that all implement the core principle of optical sectioning through spatial filtering. Different configurations exist to optimize trade-offs among spatial resolution, imaging speed, phototoxicity, penetration depth, and spectral flexibility. The major modalities of a confocal microscope: include laser scanning confocal microscopy, spinning disk confocal microscopy, multiphoton confocal microscopy, spectral confocal microscopy, and enhanced-resolution systems such as Airyscan (Figure 4). Each of these approaches modifies the illumination detection scheme to serve distinct experimental requirements.
The different types of confocal microscopy represent a spectrum of optimized imaging strategies:
- Laser scanning systems prioritize resolution and quantitative precision,
- Spinning disk systems prioritize speed and live-cell compatibility,
- Multiphoton systems enable deep tissue imaging with minimal photodamage,
- spectral systems enhance multiplexing capability, and
- Airyscan systems push resolution beyond classical limits.
The selection of an appropriate modality of a confocal microscopy is therefore fundamentally application-driven, balancing spatial resolution, temporal dynamics, photostability, and tissue penetration requirements.
Laser scanning confocal microscopy (LSCM)
LSCM represents the classical and most widely deployed implementation of confocal imaging. In this system, a highly focused laser beam is directed onto a single diffraction-limited point on the specimen. The beam is then raster-scanned across the sample using galvanometric mirrors or resonant scanners. At each position, emitted fluorescence passes through a pinhole aperture that rejects out-of-focus photons before detection, typically by a photomultiplier tube (PMT) or hybrid detector.
The key strength of LSCM lies in its high spatial resolution and optical sectioning capability. Because the system collects light from a precisely defined focal volume, it produces high-contrast images even in relatively thick or optically complex specimens. This makes it particularly suitable for fixed-cell imaging, tissue sections, and detailed structural studies where maximum resolution is prioritized over speed.
However, LSCM has inherent limitations. Since imaging is performed point-by-point, acquisition is relatively slow compared to widefield or spinning disk systems. The sequential scanning process also increases exposure time per pixel, which can lead to photobleaching and phototoxicity, especially in live-cell applications. Despite these constraints, LSCM remains the benchmark confocal modality for quantitative imaging, high-resolution z-stacks, and detailed morphological analysis.

Spinning disk confocal microscopy
Spinning disk confocal microscopy is designed to overcome the temporal limitations of laser scanning systems. Instead of scanning a single point, it uses a rapidly rotating disk (Nipkow disk) containing thousands of micro-lenses paired with corresponding pinholes. As the disk rotates, multiple excitation points are projected simultaneously onto the specimen, enabling parallelized imaging.
This architecture dramatically increases acquisition speed, making it ideal for dynamic biological processes such as vesicle trafficking, cytoskeletal rearrangements, and microbial motility. Because exposure time per point is reduced, spinning disk systems also significantly minimize photobleaching and phototoxic effects, preserving cell viability during prolonged imaging sessions.
Despite these advantages, spinning disk confocal microscopy has some trade-offs. The pinhole size and spacing are fixed by the disk design, which can limit flexibility in optical sectioning compared to adjustable pinhole LSCM systems. Additionally, because emitted light passes through multiple optical elements simultaneously, there is a modest reduction in signal efficiency, making the system less optimal for very weak fluorescence signals or deep tissue imaging. Nonetheless, it is widely regarded as the preferred platform for live-cell imaging at high temporal resolution.
Multiphoton (two-photon) confocal microscopy
Multiphoton confocal microscopy, most commonly implemented as two-photon excitation microscopy, differs fundamentally from single-photon confocal systems in its excitation physics. Instead of using high-energy visible light, it employs near-infrared femtosecond pulsed lasers. Fluorescence excitation occurs only when two lower-energy photons are simultaneously absorbed at the focal point, a nonlinear optical process. A critical consequence of this mechanism is that excitation is inherently confined to the focal volume, eliminating the need for a physical pinhole. This intrinsic optical sectioning provides excellent image contrast while significantly reducing out-of-focus excitation.
One of the most important advantages of multiphoton microscopy is its ability to image deep within scattering tissues, often reaching depths of 500-1000 µm depending on the sample. Near-infrared light experiences less scattering and absorption in biological tissue, enabling visualization of structures in intact organs, brain tissue, and thick microbial aggregates. Additionally, because excitation is restricted to the focal plane, phototoxicity and photobleaching outside the imaging region are greatly reduced, making it particularly suitable for long-term in vivo imaging. However, multiphoton systems are expensive, require high-powered pulsed lasers, and often have lower lateral resolution compared to short-wavelength confocal systems due to the longer excitation wavelength.
Spectral confocal microscopy
Spectral confocal microscopy extends conventional confocal imaging by incorporating spectrally resolved detection. Instead of collecting fluorescence through fixed bandpass filters, emitted light is dispersed using a prism or diffraction grating and recorded across a continuous wavelength spectrum for each pixel. This capability allows for spectral unmixing, a computational technique that separates overlapping emission spectra from multiple fluorophores. In complex biological samples where, fluorescent markers have similar emission peaks, spectral detection significantly improves the accuracy of signal separation and colocalization analysis.
Spectral confocal systems are particularly valuable in multiplex imaging, where multiple probes are used simultaneously to label different cellular components, microbial populations, or gene expression markers. They are also useful in samples with high autofluorescence, as background signals can often be distinguished and subtracted based on spectral signatures. The primary limitation is increased system complexity and data processing requirements, as each image contains full spectral information rather than single-channel intensity values. This results in larger datasets and more computationally intensive analysis workflows.
Airyscan and super-resolution confocal variants
Airyscan and related enhanced-detector systems represent a hybrid approach between conventional confocal microscopy and super-resolution imaging. Rather than using a single-point detector behind a pinhole, Airyscan systems employ a detector array positioned at the Airy disk plane, capturing spatially distributed emission information that would otherwise be discarded. By computationally reassessing how light is distributed across the detector elements, these systems reconstruct images with improved signal-to-noise ratio and enhanced spatial resolution. Typically, lateral resolution can be improved to approximately ~120 nm, surpassing the classical confocal diffraction limit without requiring full super-resolution techniques such as Stimulated Emission Depletion microscopy (STED) or PhotoActivated Localization Microscopy (PALM).
Airyscan technology is particularly advantageous because it maintains many benefits of conventional confocal microscopy such as optical sectioning and multicolor imaging while significantly improving resolution and sensitivity. It is especially useful for subcellular structure visualization, including membrane dynamics, protein clustering, and fine cytoskeletal architecture. However, these systems require post-processing and are more computationally intensive than standard confocal imaging. They also do not achieve the extreme resolution of true super-resolution methods, positioning them as an intermediate but highly practical enhancement.
Fluorescence and labeling in confocal microscopy
Confocal microscopy is predominantly a fluorescence-based imaging modality, relying on the excitation and emission properties of fluorophores to generate high-contrast, spatially resolved images. The technique depends on the selective labeling of specific cellular or molecular targets, enabling precise visualization of structures and dynamic biological processes within complex samples.
Fluorophores
Fluorophores are molecules capable of absorbing photons at a defined excitation wavelength and subsequently emitting light at a longer emission wavelength. This Stokes shift enables optical separation of signal from excitation light, which is essential for high-contrast imaging in confocal systems.
Common classes of fluorophores include:
- Organic dyes: Small synthetic molecules such as fluorescein, rhodamine, Texas Red, and Alexa Fluor series. These dyes are widely used due to their high brightness, photostability (especially modern derivatives), and compatibility with immunolabeling protocols. Their spectral properties can be finely tuned, making them suitable for multiplex experiments.
- Fluorescent proteins: Genetically encoded reporters such as GFP (green fluorescent protein), mCherry (monomeric Cherry fluorescent protein), CFP (cyan fluorescent protein), and YFP (yellow fluorescent protein). These proteins are expressed within living cells as fusion constructs with target proteins, enabling real-time imaging of protein localization, trafficking, and expression dynamics without the need for exogenous staining.
- Quantum dots: Semiconductor nanocrystals characterized by exceptional photostability and broad excitation spectra with narrow, size-tunable emission peaks. Their resistance to photobleaching makes them particularly useful for long-term imaging and single-molecule tracking applications, although concerns about cytotoxicity and functionalization complexity limit their routine use in live-cell systems.
Labeling strategies in confocal microscopy
Fluorescence-based confocal imaging depends heavily on how biological targets are labeled, with several widely adopted strategies:
- Immunofluorescence (antibody-based labeling): This approach uses primary antibodies that specifically bind to target antigens, followed by fluorophore-conjugated secondary antibodies for signal amplification. It is highly sensitive and widely used in fixed-cell and tissue imaging, particularly for protein localization studies.
- Genetic tagging (fusion proteins): In this method, genes encoding fluorescent proteins are fused to genes of interest, allowing endogenous expression of fluorescently labeled proteins. This strategy is especially powerful for live-cell imaging, as it preserves native cellular context and enables dynamic studies of protein behavior.
- Chemical staining: Involves the use of fluorescent dyes that bind selectively to cellular components such as nucleic acids (e.g., DAPI for DNA), membranes, lipids, or organelles. Chemical stains are simple, rapid, and effective for both live and fixed samples, although specificity may vary depending on the probe.
Multicolor imaging in confocal microscopy
Multicolor fluorescence imaging allows simultaneous visualization of multiple molecular targets within the same specimen. This is achieved by combining fluorophores with distinct excitation and emission spectra, enabling parallel detection of different structures or biomolecules. However, successful multichannel imaging requires careful experimental design to minimize spectral overlap and fluorescence bleed-through.
Key considerations in multicolor imaging include:
- Selection of fluorophores with well-separated emission peaks
- Use of appropriate filter sets or spectral detectors
- Sequential scanning to reduce channel crosstalk
- Computational spectral unmixing when overlap is unavoidable
When properly optimized, multicolor confocal imaging provides powerful insights into spatial relationships, colocalization, and molecular interactions within complex biological systems.
Optical sectioning and 3D imaging
Confocal microscopy enables true three-dimensional visualization of specimens through precise optical sectioning, allowing researchers to reconstruct volumetric structures from sequentially acquired thin optical slices. This capability is central to its use in cell biology, microbiology, and tissue imaging.
- Z-stack acquisition
A fundamental step in 3D imaging is Z-stack acquisition, where a series of optical sections are captured at incremental focal depths along the z-axis. Each image represents a discrete plane within the specimen, typically separated by step sizes ranging from approximately 0.2 to 1 µm, depending on the objective lens numerical aperture, wavelength, and required resolution. Smaller step sizes increase axial sampling fidelity but also raise acquisition time and photobleaching risk. The resulting stack forms a dense dataset representing the full depth profile of the sample.
- 3D Reconstruction
Following acquisition, specialized software reconstructs the Z-stack into a coherent three-dimensional volumetric dataset. This process involves aligning and rendering optical slices to generate spatially accurate representations of the specimen. 3D reconstruction enables detailed visualization and analysis of complex biological structures, including cellular architecture (e.g., nuclei, organelles, cytoskeletal networks), tissue organization (e.g., epithelial layering, vascular structures), and microbial assemblies such as biofilms, where spatial heterogeneity and microcolony formation are critical to function and antimicrobial resistance. Advanced rendering techniques allow rotation, slicing, and quantitative volumetric measurements.
- Deconvolution
To further enhance image quality in confocal microscopy, deconvolution algorithms are applied computationally to reduce optical blur inherent in microscopy systems. These methods use the system’s point spread function (PSF) a mathematical description of how a point source of light is distributed by the imaging system to reverse blurring effects. By iteratively reassigning out-of-focus light back to its origin, deconvolution improves both contrast and effective resolution, particularly along the axial dimension. This results in sharper boundaries, improved signal-to-noise ratio, and more accurate structural interpretation of fine biological details.
Applications of confocal microscopy
Confocal microscopy is a highly versatile imaging platform with applications spanning the life sciences, biomedical engineering, and materials research. Its ability to perform optical sectioning, generate high-contrast fluorescence images, and reconstruct three-dimensional structures makes it particularly valuable for studying complex, heterogeneous systems.
- Cell biology
In cell biology, confocal microscopy is a fundamental tool for resolving subcellular organization with high spatial precision. It enables detailed organelle visualization, including mitochondria, nuclei, endoplasmic reticulum, and Golgi apparatus, often through fluorescent tagging of specific proteins or membranes. This allows researchers to investigate organelle morphology, dynamics, and interactions under physiological and stress conditions. Confocal imaging is also widely used for cytoskeleton imaging, capturing actin filaments, microtubules, and intermediate filaments to study cell shape, motility, and intracellular transport. In addition, it is essential for protein localization studies, where fluorescently labeled antibodies or fusion proteins reveal spatial distribution patterns that are critical for understanding signaling pathways and cellular function.
- Microbiology
In microbiology, confocal microscopy is indispensable for studying spatially structured microbial systems, particularly biofilms, which are central to antimicrobial resistance and chronic infections. It allows visualization of biofilm architecture, including thickness, density gradients, and extracellular polymeric substances (EPS). It is also used to investigate microbial interactions, such as competition, symbiosis, and quorum sensing in multispecies communities. Live/dead staining assays are frequently employed to assess microbial viability within biofilms or planktonic populations, enabling quantitative evaluation of antimicrobial treatments and environmental stressors.
- Biomedical Research
Confocal microscopy plays a significant role in biomedical research by enabling high-resolution visualization of cancer cell morphology, including nuclear abnormalities, membrane changes, and cytoskeletal reorganization associated with tumor progression. It is also widely used in tissue imaging, where thin optical sections allow reconstruction of complex tissue architecture without physical sectioning. Furthermore, it supports drug distribution studies, helping researchers track fluorescently labeled compounds within cells or tissues to evaluate uptake, penetration, and intracellular localization.
- Neuroscience
In neuroscience, confocal microscopy is essential for mapping neuronal networks, allowing visualization of dendritic trees, axonal projections, and connectivity patterns. It is also used to analyze synapse structure, including pre- and postsynaptic protein localization, which is critical for understanding synaptic transmission and plasticity. In brain tissue imaging, confocal microscopy enables high-resolution reconstruction of neural circuits in fixed tissue slices, often combined with immunofluorescence labeling of neuronal markers.
- Materials Science
Beyond biology, confocal microscopy is applied in materials science for non-destructive surface characterization and structural analysis. It is used to examine surface roughness, coatings, and thin films, as well as to generate 3D topographical maps of materials. In microstructure analysis, it helps visualize grain boundaries, defects, and phase distributions in polymers, metals, and composites.
- Environmental Science
In environmental science, confocal microscopy is widely used to study soil microbial communities, enabling visualization of microbial spatial organization and interactions with soil particles. It is also critical for understanding biofilm ecology, particularly in natural aquatic and terrestrial environments where microbial consortia form structured communities. Additionally, it is used to investigate interactions with pollutants, such as tracking uptake, localization, and effects of contaminants (e.g., heavy metals, antibiotics, and microplastics) on microbial and eukaryotic cells.
References
Hibbs, A. R. (2004). Confocal microscopy for biologists. Springer.
Elliott, A. D. (2020). Confocal microscopy: Principles and modern practices. Current Protocols in Cytometry, 92(1), e68.
Callamaras, N., & Parker, I. (1999). Construction of a confocal microscope for real-time x-y and x-z imaging. Cell Calcium, 26(6), 271–279.
Castellano-Muñoz, M., Peng, A. W., Salles, F. T., & Ricci, A. J. (2012). Swept field laser confocal microscopy for enhanced spatial and temporal resolution in live-cell imaging. Microscopy and Microanalysis, 18(4), 753–760.
Dean, P. N. (1998). Confocal microscopy: Principles and practices. In Current Protocols in Cytometry (pp. 2.7.1–2.8.12). Wiley.
Discover more from Microbiology Class
Subscribe to get the latest posts sent to your email.