Diagnostic methods accurately identify viral infections in patients. This is a prerequisite to control and limit virus propagation through an effective clinical management of the disease. A precise identification of the ongoing or past viral infection helps to achieve the following:
- prevent transmission of the infection to other cells or susceptible hosts,
- set up an appropriate antiviral therapy, and
- monitor the success of ongoing antiviral treatment in the already infected individual.
Failure in diagnosis can lead to significant human and financial loss. This is why it is important to carry out appropriate viral diagnostic assay to isolate and characterize the infection virus.
Virus assays vary in approach, efficacy and cost. Besides performance parameters including accuracy, precision, sensitivity, and specificity which relate directly to the results, simplicity and ease in performing the tests are also important factors. The Turn Around Time (TAT), the interval time between sample registration and result reporting, is also used as a key performance indicator.
Examples of long-established virus assays for diagnosis
Traditional laboratory identification methods of viral infections focus mainly on optical detection of virions by microscopy techniques or by indirect serologic evidence. These long-established virus assays for diagnosis include:
a) Electron microscopy (EM)
EM for a long time has been considered an efficient tool for the direct detection of viruses by visual counting of the viral particles in body fluids, stools or histopathologic samples. Though essential in revealing morphology and appearance of the infecting virus, it requires a high virion number in samples and substantial technical skills and expertise by the operator. Typical example is an electron microscopic image of the influenza A virus subtype H1N1 shown in Figure 1.

b) Light microscopy
The analysis of viral proliferation within a cell monolayer by light microscopy is a standard diagnostic technique. Changes in cellular appearance like swelling, shrinking and syncytium formation are examined. They indicate the presence of the virus and are defined as cytopathic effect (CPE). Cytopathic effects are observable changes seen in cell morphology because of virus growth. CPE occurs because virus is present, and their presence in such environment (e.g., cell culture medium ladened with samples suspected of containing virus) usually causes morphological changes to the cells in the cell culture medium. These morphological changes in the cells are generally called CPE. Examples of CPE include:
- ballooning of cells
- death of cells
- clustering of cells
- rounding of cells (Figure 2)
Generally, Cytopathic effects are observable morphological changes that occur in cells because of viral replication. It encompasses those biochemical and/or morphological changes that occur in viral infected cells, and which indicate viral infectivity and replication in a particular host cell. When CPE occurs, the cells will lose its ability to reproduce. Although CPE is usually observed after 5–10 days of incubation, it can vary from 24 hours for herpes simplex to 10–30 days for cytomegalovirus. Incubation time for CPE is an important characteristic of a virus and can be used as a form of characterization in viral identification.

c) Fluorescence microscopy
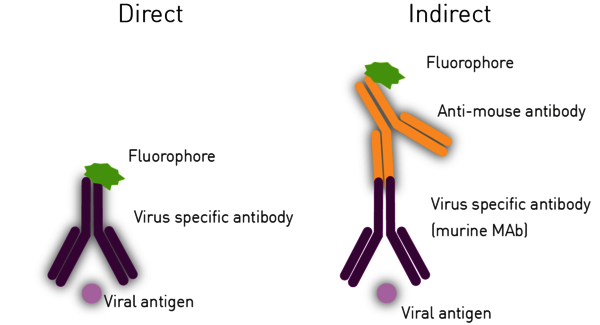
The Immuno-Fluorescence Antibody (IFA) assay detects virus in infected cells and relies on the use of specific antibodies against a viral antigen conjugated to fluorophores. The fluorophore allows visualisation of the virus distribution by fluorescence microscopy (Figure 3). Whilst it is possible to predict the type of virus using the assays previously shown, confirmatory testing such as an IFA is needed for better diagnosis.
Disadvantages of long-established microscopy virus assays
As both light and fluorescence microscopy-based virus assays rely on cell culture, they require highly trained personnel and adequate facilities. Cell culture methods are not suitable for the diagnosis of viruses which do not replicate in cell lines (norovirus, hepatitis virus) or produce CPE. Additional disadvantages include the requirement of a large volume of patient sample for inoculation and long turn around times (TATs). Therefore, these methods are often used more for research than for clinical diagnosis.
Summary of disadvantages of long-established virus assays for diagnosis
- they usually require large volume of patient samples for inoculation
- they require long TATs
- they do not support the diagnosis of viruses that do not grow or replicate in cell lines (e.g., noroviruses, hepatitis virus)
- They do not support the diagnosis of viruses that do not produce CPE
Modern techniques for the rapid diagnosis of viral infections
Whilst still employed, traditional virus assays have been or are being replaced by more rapid methods allowing qualitative testing for screening and surveillance, as well as confirmation of diagnosis of virus infections in patients. These later methods that are gradually replacing the traditional virus assay are known as modern diagnostic viral assays. They include:
- Nucleic acid amplification techniques. Examples include:
- Polymerase chain reaction
- Quantitative PCR
- Loop-mediated amplification
- Next generation sequencing
- Quantitative virus assays. Examples include:
- Plaque assay
- Focus forming assays
- Tissue culture infective dose assay
- Recombinant virus assay
- Haemagglutination assay
- Haemagglutination inhibition assay
- Enzyme linked immunosorbent assay
- Homogenous assays
Other important virus assay includes Functional virus assays – which are research and high throughput methods. These functional virus assays include:
- Neuraminidase assay
- RNA dependent RNA Polymerase assays
- Viral neutralization assay
- Viral infectivity assay
a) Nucleic Acid Amplification techniques (NAATs)
Detection of viral genetic material (DNA or RNA) in patients diagnoses infections early and is used increasingly due to its enhanced sensitivity, speed and reliability. As NAAT virus assays detect and amplify specific regions of the viral genome, they confirm viral presence even before antigens and/or antibodies appear in the bloodstream.
NAATs rely on DNA/RNA extraction from the patient sample, purification and testing for quality and concentration, followed by the selection and amplification of the genetic material of interest. The Quality assessment and quantification is commonly done by absorbance measurement using an absorbance microplate reader. Absorbance is measured at 260 nm to quantify nucleic acids and at 280 nm and 230 nm to determine contaminants post purification. For this purpose, the use of a spectrometer-based microplate reader is advantageous as it can acquire the whole absorbance spectrum of a sample, determining the concentration and purity in one reading.
Alternatively, DNA and/or RNA can be quantified using different fluorescence intensity methods like Hoechst, PicoGreen, Qubit or AccuBlue. These approaches are more sensitive as they differentiate between double-stranded and single-stranded nucleic acids. Amplification of genetic material can be achieved using various methods:
- Polymerase Chain Reaction (PCR): PCRamplifies the copy number of a viral DNA region by using virus-specific primers and a thermostable DNA polymerase. A fluorescent probe binds the amplified target and emits a detectable signal. PCR cannot be used to directly detect RNA viruses and so a Reverse Transcription PCR (RT-PCR) of the RNA genome into DNA is required prior to performing the amplification reaction.
- Quantitative PCR (qPCR): qPCRincludes a fluorescent quantification step after each amplification cycle. Once fluorescence intensity rises above background, the detectable level proportionally corresponds to the initial number of template DNA molecules in the sample. A standard curve allows the determination of the absolute quantity of target virus DNA or transcribed RNA.
- Loop-mediated Amplification (LAMP) assay: LAMP assayis a thermostable amplification reaction at 65ºC. Unlike PCR, it does not rely on temperature cycling, and can be hence measured in real-time in a microplate reader with periodic shaking. This is demonstrated by the example of SARS-CoV-2 in the application note LAMP assay for detecting SARS-CoV-2 RNA using an absorbance-based colorimetric readout. The assay can be colorimetric or fluorescent. Compared to PCR, this approach has speed and cost advantages. In addition, it is highly automatable, making it one of the best virus assays for rapid diagnosis.
- Next Generation Sequencing (NGS): NGS is a generic term to describe different modern DNA/RNA sequencing technologies that offer a much quicker and less expensive approach than first generation Sanger sequencing. These methods can provide important information on novel viruses to assist in the production of vaccines and treatments against them. The accurate quantification and purity check of DNA or RNA prior to running NGS is critical to the success of the procedure.
NAATs are very useful in virus detection. However, they can neither assess its ability to replicate and infect cells, nor detect past infections or acquired immunity. To achieve these answers, quantitative infectivity and serological tests are required. Both quantitative infectivity and serological tests are explained in this section.
b) Quantitative virus assays
Virus quantification is crucial for research and for production of vaccines, viral antigens, viral recombinant proteins, or antiviral agents. Quantitative infectivity assays determine the capability to enter the cell and utilise its molecular replication machinery. Virus infectivity assays estimate the virus concentration and can be performed in cell culture or using egg infection. In cell culture, a titration of virus is applied to a constant number of cells and given time to invade and replicate. Infectivity is then quantified using a variety of virus assays.
- The plaque assay: This assayrepresents the gold standard and most used quantitative virus assay. In plaque assays, a confluent monolayer of cells is infected with unknown concentrations of a lytic virus at varying dilutions. Infected monolayers are then covered with an immobilizing overlay medium to prevent viral spread. Zones of cell death (plaques) will begin to develop as result of constrained infection and replication. Infected cells will propagate the replication-lysis-infection cycle, resulting in increasingly distinct and discrete plaques (Figure 4). Visible plaque formation can take 2–14 days, depending on the virus and host cells used. Plaques are typically counterstained by neutral red or crystal violet to be counted manually. In combination with the dilution factor used, the viral titre is calculated in terms of plaque forming units (PFU) per millilitre (PFU/ml). The pfu/ml represents the number of infective particles within the sample and assumes that each plaque formed is representative of one infective virus particle.

- Focus Forming Assays (FFAs): FFAsare modified plaque assays that utilise an antibody-based staining method to detect infected cells. Unlike plaque assay, FFAs detect lytic and non-lytic viruses with increased sensitivity and decreased incubation times. FFAs are limited in their need for appropriate antibodies and their ability to only probe viral protein subunits and not infectious virions. The results of focus forming virus assays are expressed in focus forming units (FFU) per millilitre (FFU/ml).
- The 50% Tissue Culture Infective Dose (TCDi50) assay: TCDi50 assayquantifies the amount of virus required to produce CPE in 50% of inoculated cells. It is an endpoint dilution assay used to quantify viruses that do not form plaques. In brief, serial dilutions of the virus are added to host cells. After incubation, the percentage of cell death is measuredfrom a cell viability assay using a plate reader. Alternatively, microscopy can be used. The dilution at which 50% of the cell cultures are infected (the end point) is used to mathematically calculate a TCID50 result and is expressed as 50% infectious dose (ID50) per millilitre (ID50/ml).
- Recombinant virus assay: Recombinant virus assay can be engineered to express a fluorescent or luminescent marker. This virus assay detects virions faster than plaque assays or TCID50 and is ideally suited to screen for antivirals. Additionally, it can be run on a microplate reader allowing automation. Samples containing unknown quantities of luciferase-expressing or fluorescence-expressing viruses are transferred on to a plaquing cell line in parallel with the standard dilution curve. The luminescence or fluorescence is read after incubation and viral titres are calculated based on the standard curve.
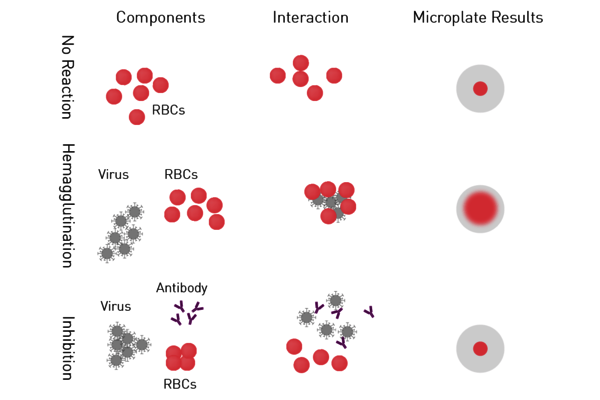
- Hemagglutination assay and Hemagglutination Inhibition Assay (HIA): Haemagglutinin is a protein on the envelope of arboviruses, influenza and parainfluenza virus subtypes which binds red blood cells (RBCs) to form a lattice of agglutinated cells. In the hemagglutination assay, serial dilutions of the virus are added to RBCs. Samples are then screened for agglutinated cells (Figure 5). A variation of this virus assay is the hemagglutination inhibition assay used to measure specific antibodies in serum. If antibodies are present in the serum at a sufficient concentration, they will interfere with the viral attachment to RBCs, resulting in inhibition of hemagglutination. Both virus assays provide a relative virus quantitation.
- Enzyme-Linked Immunosorbent assays (ELISA): ELISA measure the presence and/or concentration of antibodies generated by an infection or viral antigens in serological samples. As antibodies persist in the bloodstream even for many years after infection, a positive test is not an indicator of active infection but determines patient immunity resulting from exposure to the virus, reinfection, or a reactivation state. ELISAs are invaluable for studying the epidemiology of disease as they study present and past disease prevalence in different populations over time.
ELISAs essentially employ a viral antigen or antibody against the pathogen immobilised on the surface of a microplate well. This is recognised and bound respectively by viral antibodies or specific proteins of the virus present in the samples added to the well.
Specific labelled antibodies recognise the bound molecule of interest and produce a colorimetric (absorbance), chemiluminescent or fluorescent signal that is detected by a microplate reader. The signal is proportional to the amount of specific antigen or antibody in the sample.
For indirect ELISAs, detection involves a two-step process whereby a non-labelled antibody binds to the antigen or viral antibody and is then recognised by a labelled secondary antibody (Figure 6).
ELISAs are also used in a competitive way, measuring the concentration of an antigen by detection of signal interference. The sample antigen competes with a reference antigen for binding to a specific labelled antibody. The reference antigen is pre-coated on a multi-well plate. The sample is pre-incubated with labelled antibody and added to the wells. Depending on the amount of antigen in the sample, the free antibodies will be available to bind the reference antigen. This means the more antigen there is in the sample, the less reference antigen will be detected and the weaker the signal. This method can also be adapted to detect antibodies in samples.

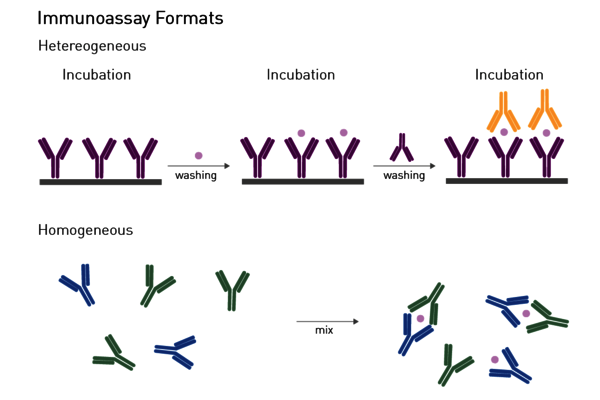
Homogeneous assays: Homogenous assays are an alternative to traditional heterogeneous ELISAs (Figure 7). Unlike ELISAs, removal of the unbound components from the well is not required to reduce the background and detect the bound complex. Hence, homogeneous assays do not necessitate in-between separation or washing steps and can be executed with a simple add-and-read protocol. This minimises handling steps, operation time and makes them particularly suited for automation-supported screening. Homeogenous virus assays are fluorescence polarization immunoassays, AlphaScreen®/AlphaLISA® technologies, and different TRF and TR-FRET based assays.
Functional virus assays: research and high throughput methods
There are different types of functional assays to study how a virus works, such as how it infects cells and which mechanisms and enzymes are important for the virus. Functional methods are employed for both: basic research as well as for high throughput screening (HTS) drug screening.
- Methods in basic research: in basic research, functional virus assays help to find immunogenic components for vaccines and potential drug targets for therapy. Microplate-based tests employed for virus characterization, mainly use plates up to 96 wells. As soon as a virus is understood and potential drug targets are identified, higher throughput virus assays search for molecules interfering with pathogenic processes.
- High throughput assays: the development of antiviral agents requires to screen compound libraries for various aspects and relies on high throughput compatible virus assays. These are performed in plate formats of 384 wells or 1536 wells. In addition to the plate density, high throughput virus assays are automatically prepared with the use of automated liquid handlers and industrial robots.
Neuraminidase assay
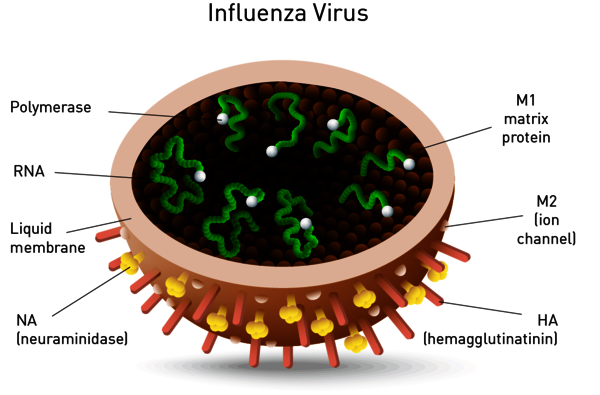
Neuraminidase is one of two key glycoproteins found on the surface of influenza viruses (the other being hemagglutinin, Figure 8). Neuraminidase is crucial to the release of influenza from infected cells and enables spread within the respiratory tract. Viral neuraminidase cleaves sialic acid groups from glycoproteins on the host cell surface, leading to release of virus particles from infected cells. It is therefore the target of new therapies such as the FDA-approved neuraminidase inhibitors zanamivir and oseltamivir. Its activity further reports on viral titer and potential neuraminidase inhibitor resistance.
Neuraminidase activity and inhibition is mainly measured using a synthetic substrate which releases a fluorophore, if processed by the enzyme. In this assay, the virus is mixed with the substrate and fluorescence intensity is measured on a microplate reader. The intensity is directly related to neuraminidase activity. A fluorescence-based neuraminidase assay was measured by Hong Kong-based researchers on the CLARIOstar microplate reader and revealed a neuraminidase inhibitory activity by traditional multi-herb formulations.
A second type of neuraminidase assay employs chemiluminescence instead of fluorescence and provides higher sensitivity. This virus assay uses a sialic acid derivative which emits light in the presence of active neuraminidase.
RNA dependent RNA Polymerase assays
Another target for RNA viruses such as severe acute respiratory syndrome (SARS, hepatitis C and influenza is viral RNA-dependent RNA Polymerase (RdRP) (Figure 9). Viruses localized in the cytoplasm of a cell do not have access to the host’s RNA polymerase and encode their own polymerase to allow replication. The RdRP enzyme transcribes RNA from a viral RNA template, in contrast to DNA-dependent RNA polymerase that transcribes RNA from the host DNA template. RdRP enzyme activity and inhibition are important virus assays and rely on multiple RNA-dependent RNA polymerase assays.
RNA transcribed by RdRP is measured by fluorescent RNA dyes that increase their fluorescence in the presence of RNA. Recombinant RdRP or RdRP isolated from infected cells catalyses in vitro RNA synthesis. The increase in RNA is then measured using RNA dyes such as SYTO-9, Quantifluor or a protein-based sensor.
Measuring nucleotide incorporation is a traditional virus assay to determine the activity of RdRP by in vitro RNA transcription. The RdRP enzyme is mixed with a biotin-labelled primer to initialize the reaction and labelled nucleoside triphosphates (NTPs). If the enzyme is active, it elongates the primer strand and incorporates the labelled NTPs. The complex of primer and newly synthesized RNA is subsequently immobilized on a streptavidin coated microplate and excessive NTPs can be washed away. Dependent on the activity of RdRP, the amount of NTPs immobilized onto the microplate differs. In the past, the NTPs were radioactively labelled. Today fluorophores are used and quantified in a fluorescence microplate reader.
A fluorescence polarization-based (FP) assay also uses the in vitro complementation of a template RNA strand by RdRP to determine its activity. A single stranded “template” RNA is fluorescently labelled and displays a low FP value. If RdRP synthesizes the complementary strand, the molecule enlarges its mass (double-stranded) leading to an increase in FP.

Viral neutralization assays
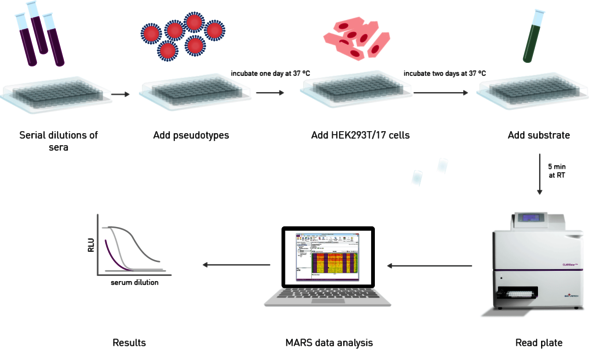
The effectiveness of an antibody, vaccine treatment or infection-related immunity is tested with a neutralization assay. The purpose of this virus assay is to evaluate the ability of an antibody to bind viral surface antigens that are required for a successful viral replication thus neutralizing the infection. The neutralization assay combines incubation of the virus or pseudotypes with samples containing potentially neutralizing antibodies, e.g. sera of vaccinated humans/animals or therapeutics. This is followed by a cell-based infection assay, which provides information as to whether the virus can infect the cells despite treatment. Neutralization assays were traditionally performed with a virus solution mixed with the neutralizing samples at different concentrations. Due to the infection risk of using the native pathogen, today, pseudotypes are used. Pseudo viruses are chimeric viruses bearing the viral glycoproteins of interest without the disease-causing phenotype. For a neutralization assay, they are engineered to express reporter genes for luciferase or GFP that are activated in the infected cell when undergoing replication. This way, the neutralization assay is easily detected in a fluorescence or luminescence microplate reader (Figure 10).
Virus infectivity assays to study antiviral activity
Methods used to test the antiviral effect of compounds overlap with assays used to diagnose infections. For in vitro and high throughput assays, cell-based microplate assays are employed.
Studying the cytopathic effect of a virus in presence and absence of a compound identifies if it prevents infection. Infectivity assays to detect cytopathic effects in screening are mainly based on measuring viability with an ATP-dependent luciferase such as CellTiter-Glo® or ToxiLightTM. These assays are compatible with a 1536 well format and reagents can be added directly to cells after the appropriate incubation of cells, virus and compound. The microplate is subsequently measured on a high throughput luminescence plate reader where low signals correlate to a high cytopathic effect. In addition to a toxic effect related to infection, this assay reports on toxicity induced by the compounds themselves in control wells with compound and cells lacking the virus.
Source:
www.bmglabtech.com/virus-assays/
Discover more from Microbiology Class
Subscribe to get the latest posts sent to your email.